Introduction
Atrioventricular septal defect (AVSD) is a congenital cardiac malformation characterized by incomplete atrial and ventricular septa development and abnormalities of the atrioventricular valves (AVVs), resulting in a common or partially separate atrioventricular (AV) orifice.[1] AVSD encompasses a spectrum of defects, ranging from partial to complete forms. Partial AVSD typically presents with an ostium primum atrial septal defect, separate AVVs with a common junction, an inlet ventricular septal defect, and a cleft mitral valve. In contrast, complete AVSD features a common AVV, an ostium primum atrial septal defect, and an unrestricted inlet-type ventricular septal defect.[2]
AVSD accounts for approximately 4% to 5% of all congenital heart defects and is strongly associated with chromosomal abnormalities, particularly trisomy 21 (Down syndrome). The condition is a leading cause of early heart failure in infants. However, long-term outcomes after surgical repair have significantly improved due to advances in diagnostic techniques, surgical strategies, and perioperative care. This course provides a comprehensive overview of AVSD, including its embryology, epidemiology, pathophysiology, clinical presentation, diagnostic evaluation, management strategies, complications, and overall clinical significance.
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
AVSD arises from abnormal development of the endocardial cushions during early embryogenesis, specifically between the fifth and eighth weeks of gestation. The endocardial cushions are critical for forming the lower atrial septum, upper ventricular septum, and AVVs. Failure of these structures to fuse properly results in the characteristic atrial and ventricular septal defects and AVV abnormalities seen in AVSD (see Image. Anatomy of a Heart with Atrioventricular Septal Defects).
Genetic mutations are the primary cause of AVSD, with most cases associated with syndromic conditions. Down syndrome (trisomy 21) has the strongest correlation, with approximately 1 in 6 individuals affected by both conditions. The Down syndrome cell adhesion molecule gene has been implicated in the pathogenesis of AVSD and other congenital heart defects in individuals with Down syndrome.[3][4] Additionally, AVSD can be associated with other genetic syndromes, including CHARGE (coloboma, heart defects, atresia of the choanae, retardation of growth/development, genital hypoplasia, and ear anomalies/hearing loss) syndrome, Ellis-van Creveld syndrome, Smith-Lemli-Opitz syndrome, and 3p deletion syndrome. Beyond syndromic associations, AVSD can also occur due to autosomal dominant gene mutations in nonsyndromic cases.
Although less commonly implicated, environmental factors have been associated with increased risk of AVSD. Gestational diabetes and maternal obesity are recognized contributors to nonsyndromic AVSD development.[5] Furthermore, AVSD may occur in the context of complex congenital heart disease and heterotaxy syndromes, where abnormal left-right axis development affects cardiac anatomy. In summary, AVSD results from defective endocardial cushion development, most often due to genetic mutations associated with syndromic conditions, particularly Down syndrome, but also involving other genetic syndromes and environmental risk factors.
Epidemiology
AVSD accounts for approximately 3% to 7% of all congenital cardiac malformations, with an estimated incidence of 0.24 to 0.31 per 1000 live births.[2][6] While both sexes are affected, some study results suggest a slight female predominance, particularly in patients with Down syndrome, with a female-to-male ratio of 1.3 to 1.0.[7] The association with trisomy 21 is significant, with 40% to 50% of children with Down syndrome having an AVSD, making it the most common congenital heart defect in this population.
Data from the Society of Thoracic Surgeons congenital database reported 4138 cases of complete AVSD between 2013 and 2017 (see Image. Diagnosed Cases of Atrioventricular Septal Defects). Most underwent surgical repair with low mortality rates (2.6% to 2.9%) when treated with or without valvuloplasty (see Image. Atrioventricular Septal Defect Procedural Case Numbers). However, cases requiring valve replacement demonstrated significantly higher perioperative mortality (16.7%).
Notably, intraoperative conversions from failed valvuloplasty to valve replacement had a mortality rate as high as 50%, underscoring the critical importance of accurate preoperative imaging and surgical planning to optimize outcomes (see Image. Operative Mortality of Atrioventricular Septal Defect Procedures). The widespread use of prenatal ultrasound and advances in congenital heart disease screening continue to influence reported incidence rates. Still, AVSD remains a common and clinically significant congenital defect, especially in populations with chromosomal abnormalities.
Pathophysiology
Endocardial cushions are paired (superior and inferior) mesenchymal structures located in the common AV canal in the early embryonic period, and their growth is of prime importance in the development of the AV septum and AVVs.[8] These endocardial cushions fuse at the end of the fourth week of development and form 2 atria and 2 ventricles. Failure of fusion results in a variable degree of AVSD.
The endocardial cushions, derived from the nonchamber myocardium of the AV canal, serve as foundational mesenchymal structures in the embryologic formation of cardiac valves and septa. Their morphogenesis relies heavily on signaling pathways driven by vascular endothelial growth factor, and variants within genes regulating this pathway have been implicated in congenital malformations affecting the AVVs and septa.
The notably elevated incidence of AV canal anomalies in individuals with trisomy 21 provides insight into the genetic underpinnings of endocardial cushion development. A longstanding hypothesis attributes this susceptibility to enhanced cellular adhesiveness observed in trisomy 21-derived cells. Experimental evidence supports this, as fetal fibroblasts from trisomy 21 endocardial-cushion regions exhibit increased in vitro adhesion compared to those from euploid counterparts. If endocardial cushion fusion is contingent on precise temporal and spatial parameters, then delayed cellular migration within the trisomic embryo may significantly disrupt this critical process.[9]
Collagen type VI, particularly its alpha-1 and alpha-2 chains encoded by the COL6A1/COL6A2 gene cluster located in the congenital heart disease–critical region of chromosome 21, is thought to contribute to this pathological mechanism. Fibroblasts from individuals with Down syndrome demonstrate altered adhesion properties to type VI collagen, further implicating matrix interactions in the etiology of these defects. Despite the presence of trisomy, only approximately 50% of individuals with Down syndrome manifest structural heart disease, prompting investigations into genetic modifiers both within and outside the trisomic chromosome that may influence the penetrance of AV canal defects.[10][11]
Enrichment of pathogenic variants has been observed in multiple genes—VEGF-A, COL6A1, CRELD1, FBLN1, FRZB, GATA5, NOTCH4, and CEP290—among individuals with Down syndrome and AV canal anomalies, suggesting a multifactorial and genetically heterogeneous basis for these defects.[12] Beyond syndromic associations, familial clustering of AV canal anomalies with an autosomal dominant inheritance pattern and incomplete penetrance has been documented, highlighting monogenic contributions. To date, 5 loci have been definitively associated with nonsyndromic AVSDs, designated AVSD1 through AVSD5:
- AVSD1 maps to chromosome 1p31–p21.
- AVSD2 is attributed to pathogenic mutations in CRELD1 on chromosome 3p25.
- AVSD3 involves GJA1, encoding connexin 43 (Cx43), on chromosome 6q22.
- AVSD4 is linked to GATA4 on chromosome 8p23.1.
- AVSD5 is associated with GATA6 mutations on chromosome 18q11.1.
The CRELD protein family, comprising matricellular proteins implicated in cell-cell interactions, plays a role in these pathologies. Among individuals with AVSDs unrelated to trisomy 21, approximately 6% harbor coding-region missense mutations in the CRELD1 gene. The GJA1 gene encodes connexin 43, a gap junction protein highly expressed in the ventricular myocardium and cardiac neural crest derivatives; compound heterozygous mutations have been reported in cases of AVSD.[13][14]
The GATA family of transcription factors, known for orchestrating lineage-specific gene expression, also contributes to cardiac morphogenesis. Neural crest-derived ectomesenchymal cells, originating in the cranial neural folds, are crucial for outflow tract septation and the morphogenesis of branchial arch structures. These cells navigate through pharyngeal arches 3, 4, and 6 to participate in partitioning the conotruncus and aortic sac. Experimental ablation of preotic neural crest populations in animal studies has resulted in a range of conotruncal anomalies, including truncus arteriosus and subarterial ventricular septal defects, underscoring the indispensable role of these cells in cardiovascular development.[14]
Recent research has identified the SOX7 gene as a novel pathogenic candidate in the etiology of AVSD. SOX7 appears to modulate the endothelial-to-mesenchymal transition (EndMT), which is crucial for AV cushion formation, by influencing the WNT4–BMP2 signaling pathway. Specifically, SOX7 deficiency leads to the downregulation of WNT4 expression in the endocardium, which diminishes BMP2 expression in the adjacent myocardium. This disruption impairs the EndMT process, ultimately contributing to the pathogenesis of AVSD.[15]
Classification
AVSDs represent a spectrum of congenital anomalies characterized by varying degrees of atrial and ventricular septal involvement, as well as abnormalities of the AVVs. Based on AVV morphology and its development, AVSDs are classified into partial, intermediate, and complete. These 3 anatomical variants represent a phenotypic continuum resulting from a shared developmental defect at the level of the endocardial cushions, reflecting a common genetic and embryologic basis.[16][17]
In the partial form of AVSD, also termed partial AV canal, the defining feature is an atrial septal defect located immediately adjacent to the AVV plane. This defect typically appears as a crescent-shaped gap in the lower segment of the atrial septum and is associated with a morphologically trifoliate left AVV, which frequently results in variable degrees of valvular regurgitation. Although historically referred to as an ostium primum atrial septal defect, this terminology is anatomically misleading, as the lesion does not originate from the septum primum.
The intermediate or transitional subtype of AVSD occupies a position between the partial and complete forms in terms of structural complexity. This is characterized by the presence of 2 distinct AVV orifices—right and left—along with a premum-type atrial septal defect and a ventricular septal defect situated beneath the AVV plane. The ventricular component of the defect is typically localized to the inlet portion of the septum. Unlike in complete AVSD, there is no exposed or "bare" crest at the superior margin of the interventricular septum in these cases.
In complete AVSD, also called complete AV canal defects, the malformation comprises interatrial and interventricular communication beneath a single, undivided common AVV. This valve bridges the ventricular septum, incorporating both superior (anterior) and inferior (posterior) bridging leaflets. A characteristic feature of this form is the presence of an uncovered or "bare" zone at the crest of the ventricular septum, representing the common atrioventricular junction.
In the complete form of the AVSD, a common AVV has 5 leaflets, including superior bridging, inferior bridging, left mural, right mural, and anterosuperior. Rastelli divided complete AVSD into 3 anatomical subgroups (A, B, and C), based solely on the degree of attachment and bridging of the superior leaflet to the ventricular septum.[18] These subgroups are as follows:
- Type A
- The superior bridging leaflet is equally divided and attached to the interventricular septum (69% incidence) (see Image. Atrioventricular Septal Defect, Rastelli Type A).
- Type B
- This type features aberrant papillary muscle insertion from the right septum to the left bridging leaflet (9% incidence) (see Image. Atrioventricular Septal Defect, Rastelli Type B).
- Type C
- The superior bridging leaflet is undivided with no septal attachment, leaving a large defect (22% incidence) (see Image. Atrioventricular Septal Defect, Rastelli Type C).
Rastelli type A is associated with left-sided obstruction, type B is the least common form of the complete AVSD, and type C is associated with tetralogy of Fallot and other complex congenital heart diseases.[19]
The Concept of Balance in AVSDs
Approximately one-tenth of AVCDs exhibit disproportionate commitment of the common AVV to 1 ventricle, resulting in an unbalanced AVCD. In these cases, the AVV preferentially directs inflow toward the right or left ventricle, resulting in right-dominant or left-dominant anatomical configurations, respectively. This imbalance is closely associated with underlying ventricular size and development disparities, with unequal AVV inflow serving as a cardinal diagnostic feature.
Right ventricular (RV) dominance is observed approximately twice as often as left ventricular (LV) dominance. RV dominance frequently presents alongside hypoplasia of the LV, a parachute-like configuration of the left AVV, closely approximated LV papillary muscles with a diminutive or absent mural leaflet, and may coexist with other structural anomalies such as a bicuspid aortic valve or coarctation of the aorta. In contrast, AV canal defects with LV dominance are typically accompanied by RV hypoplasia and obstructive lesions of the right ventricular outflow tract, including pulmonary stenosis or pulmonary atresia.
Efforts to objectively assess the degree of AVV and ventricular imbalance have led to the development of various echocardiographic indices aimed at stratifying patients for biventricular versus univentricular surgical strategies. Among these tools is the atrioventricular valve index—which quantifies the relative area of the AVV committed to each ventricle—as well as assessments of the angular alignment of AVV inflows and measurements relating valve inflow dimensions to the size of the ventricular septal defect. Despite their theoretical utility, these metrics often suffer from limited reproducibility and have not gained widespread clinical adoption.
In recent years, advanced imaging modalities such as cardiac magnetic resonance and electrocardiogram-gated cine computed tomography have emerged as valuable adjuncts for detailed anatomical assessment. These techniques offer superior spatial resolution and enhanced accuracy in quantifying ventricular volumes and AVV geometry, thus playing an increasingly critical role in preoperative planning for patients with unbalanced AV canal anatomy.[20] Ultimately, AVSD represents a genetically and anatomically heterogeneous condition, where disrupted development of the endocardial cushions underlies its pathophysiology. The manifestation of AVSD depends on a complex interplay of genetic mutations, chromosomal anomalies such as trisomy 21, and environmental factors, all of which influence the timing and integrity of cushion fusion, septation, and valve formation.
History and Physical
Clinical presentation of AVSDs is influenced by the type of AVSD, the magnitude of the intracardiac shunt, and other associated cardiac malformations.[6] In patients with complete AVSD, signs of pulmonary congestion and right heart failure develop in early infancy due to a significant left-to-right shunt as pulmonary vascular resistance drops after birth. Heart failure and Eisenmenger may develop even earlier if these patients have associated AVV regurgitation, ventricular imbalance, or coarctation of the aorta.[21]
Patients with a partial AVSD without other complex congenital cardiac malformations and minimal AVV regurgitation usually remain asymptomatic in infancy and early childhood. They are diagnosed based on incidental findings, including pulmonary or tricuspid flow murmur and a fixed splitting due to an atrial septal defect. Symptoms of heart failure in infants may include difficulty in feeding, sleepiness, lethargy, and failure to thrive, while children may have a complaint of dyspnea.
Signs of heart failure include:
- Tachypnea, tachycardia
- S3 gallop
- Rales on chest auscultation
- Raised jugular venous pressure
- Tender hepatomegaly
- Wide fixed splitting due to an atrial septal defect
- Pansystolic murmur due to AVV regurgitation
- Pulmonary flow murmur due to increased flow through the pulmonary valve
- Middiastolic flow murmur due to increased flow through the tricuspid valve
General physical examination may reveal cyanosis when there is a reversal of shunt (Eisenmenger syndrome) and dysmorphic features in case of associated syndromes.
Evaluation
Antenatal Evaluation
Antenatal ultrasonography with a 4-chamber view is the commonly used diagnostic test for AVSD. The most common findings include a common AVV and a defect in the atrial or ventricular septum. However, the sensitivity of antenatal ultrasound for the AVSD is very low.[22]
Postnatal Evaluation
The postnatal evaluation of patients with atrioventricular septal defects or related anomalies involves several key imaging and diagnostic modalities. Chest radiography typically shows cardiomegaly and pulmonary plethora, particularly in cases with significant AVV regurgitation. The electrocardiogram reveals characteristic features, including a superior axis in the frontal plane, RV hypertrophy, and varying degrees of AV block. Additional findings may include a superior P wave axis and partial right bundle branch block.
Echocardiography remains the primary tool for detailed anatomical and functional assessment, demonstrating abnormalities such as:
- Abnormal configuration of the AVV
- Loss of the normal offset of the AVV
- Abnormal position of the papillary muscles
- Disproportion in the LV inlet and outlet
- Ostium primum atrial septal defect
- Ventricular septal defect of inlet type
- Other associated cardiac malformations [23]
Cardiovascular magnetic resonance imaging provides complementary and often more precise evaluation, particularly for measuring defect sizes and quantifying the regurgitation fraction across the AVV, supporting comprehensive preoperative planning and management.
Treatment / Management
The management of AVSDs can be divided into medical and surgical treatment.[24]
Medical Treatment
This includes diuretics and vasodilators to reduce preload and afterload, thereby relieving pulmonary congestion and heart failure symptoms. Associated feeding problems and failure to thrive are managed by tube feeding and providing extra calories. In AVSDs, medical treatment is typically aimed at optimizing the patient's condition for surgery.[6]
Surgical Treatment
Surgical correction is the ultimate treatment for AVSD. AV septal repair is a complex surgical procedure and carries an operative mortality of more than 3% even in the contemporary era of advanced surgical techniques.[25] This procedure also carries significant postoperative mortality and morbidity due to residual intracardiac shunts, AVV regurgitation, left ventricular outflow tract obstruction, and arrhythmias.[26]
The assessment of preoperative imaging and hemodynamic data is crucial for selecting the optimal surgical procedure, reducing the need for recurrent surgery and postoperative complications.[27] In previous studies' results, the requirement for recurrent procedures has been reported as high as 18.2% at 15 years after surgical correction. Left AVV dysplasia, absence of cleft closure, and associated cardiac malformations have been found to increase the rate of recurrent procedures.[28](B2)
Surgical Approaches for Repairing Complete AVSDs
The operative correction of complete AVSDs may be undertaken using 1 of several established techniques, each aimed at reconstructing the AV septum while preserving valvular function and minimizing postoperative complications. These techniques include:
- Single-patch repair
- This method involves using a single patch to close the defect's atrial and ventricular septal components simultaneously. Following the division of the common AVV, the right and left valve components are reattached to the newly created septal structure. Closure of the left AVV cleft is routinely performed as part of this reconstruction. Untreated autologous pericardium is commonly utilized as the patch material. Careful attention is given to anchoring the AVV tissue close to the crest of the ventricular septum, which facilitates optimal leaflet coaptation and function.
- Two-patch repair technique
- This approach employs separate patches to individually address the atrial and ventricular septal defects. Typically, a Dacron patch is used to close the ventricular component, while the atrial septum is reconstructed with a patch fashioned from autologous pericardium. As with the single-patch technique, the cleft in the left AVV is closed primarily. The ventricular patch must be undersized in the anteroposterior axis and maintain a low patch profile to prevent excessive displacement of valve tissue into the atrial chamber, which has been correlated with an increased risk of postoperative left AVV regurgitation.
- Modified single-patch ("Australian") technique
- In this variation, the ventricular component is eliminated by directly suturing the common AVV leaflets to the crest of the ventricular septum. A separate pericardial patch is then used to close the primum atrial septal defect. Closure of the left AVV cleft is carried out similarly to the techniques mentioned above.
All 3 strategies have demonstrated favorable early outcomes; however, none eliminates the possibility of late reoperation, particularly in residual or recurrent left AVV regurgitation. The likelihood of requiring reintervention appears to be higher in patients presenting with significant preoperative valve insufficiency. Accordingly, comprehensive echocardiographic assessment is imperative before hospital discharge and throughout long-term follow-up.[29][30][31]
In complete AVSD, surgical closure should be performed in early infancy to reduce pulmonary vascular disease. In incomplete AVSD, a repair can be slightly delayed if the patient is not symptomatic. For partial AVSD, the primary repair is preferred, typically involving patch closure and AVV valvuloplasty.
For balanced complete AVSD, early primary repair with 2-patch closure techniques is preferred over 1-patch closure, as 1-patch closure is associated with an increased rate of recurrent procedures due to patch dehiscence and residual shunt. Pulmonary artery banding is no longer a routine procedure to repair complete AVSD. For unbalanced complete AVSD, the repair technique may include single-ventricle palliation with staged or primary biventricular repair.[26]
Differential Diagnosis
Common differential diagnoses of AVSD include ostium secundum atrial septal defect, isolated ventricular septal defect, and tetralogy of Fallot. The symptoms of heart failure and enlargement of cardiac chambers are common in these malformations, and an echocardiogram plays a major role in differentiating AVSD from the aforementioned conditions.
Prognosis
The prognosis of untreated AVSD is dismal. Around 50% of the patients die during infancy, either due to heart failure or pulmonary infections.[2] Those who survive beyond 1 year develop irreversible pulmonary vascular disease, and later on, the reversal of the shunt. Patients undergoing surgical repair have a 15-year survival rate of approximately 90%, and 9% to 10% of those require reoperation within this timeframe.[32]
Complications
Most of the complications of AVSD are related to intracardiac shunts or AVV regurgitation. In complete AVSD, shunting of blood from left to right leads to right-sided overload and signs of heart failure and pulmonary congestion at a very early age, which contributes to significant mortality during infancy. If the shunt is not corrected, it causes an irreversible pulmonary vascular disease that leads to pulmonary hypertension and Eisenmenger syndrome.
Regurgitation of blood from the ventricle to the atria through the AVV leads to pulmonary congestion and enlargement of the atrium. Enlargement of the atrium can lead to supraventricular arrhythmias. Other complications are related to poor feeding, which may include malnutrition and failure to thrive.
Deterrence and Patient Education
Deterrence strategies for AVSD primarily focus on preventing risk factors and early detection, particularly during the prenatal or perinatal period. Since AVSD is often associated with genetic syndromes—most notably trisomy 21—genetic counseling is a critical component of deterrence. Parents with a family history of congenital heart disease or known chromosomal abnormalities may benefit from genetic screening and counseling before or during pregnancy. Additionally, good prenatal care, avoidance of teratogenic substances, effective control of maternal diabetes, and routine fetal anomaly scans (such as second-trimester fetal echocardiography) can aid in the early identification and informed planning of perinatal care. However, primary prevention of AVSD is often limited due to its primarily congenital and usually genetic origin.
Patient education is essential throughout the continuum of care, from diagnosis to long-term management. For parents of children diagnosed with AVSD, especially in the prenatal or neonatal period, education should address the nature of the defect, its clinical implications (eg, heart failure, failure to thrive, and pulmonary overcirculation), and the need for timely surgical intervention. Emphasis should be placed on understanding the difference between partial, transitional, and complete AVSD, as this influences the treatment approach and prognosis.
Families should be counseled about the surgical options, expected outcomes, and the importance of early repair to prevent irreversible pulmonary vascular disease. Education should also include potential complications such as residual septal defects, arrhythmias, AVV regurgitation, or the need for reoperation. For patients with AVSD associated with Down syndrome, caregivers should be informed about the increased surgical complexity and specific postoperative considerations in this population, including the potential for persistent pulmonary hypertension or atrioventricular valve dysfunction.
Long-term follow-up with pediatric cardiology is critical, and families should be encouraged to adhere to scheduled evaluations, including echocardiography, electrocardiograms, and clinical assessments. As patients transition into adolescence and adulthood, education should focus on ongoing cardiac surveillance, endocarditis prophylaxis in specific situations, and the condition's impact on pregnancy and physical activity. Overall, comprehensive and age-appropriate patient and family education fosters informed decision-making, improves adherence to treatment and follow-up, and enhances quality of life.
Pearls and Other Issues
Diagnosis of AVSD in fetal life or the early neonatal period is essential to initiate appropriate medical treatment and to plan early surgical repair. In the contemporary era of advanced surgical techniques, the operative mortality of AVSD repair is low, with excellent long-term outcomes even in patients with Down syndrome. Atrioventricular valve regurgitation is the most common reason for reoperation; therefore, assessing the imaging data before surgical repair and paying close attention to AVV repair during the primary repair is essential. Postoperatively, patients should be followed regularly for AVV regurgitation and left ventricular outflow tract obstruction.[6]
Enhancing Healthcare Team Outcomes
Optimal care for patients with AVSD requires a coordinated, multidisciplinary approach that begins at diagnosis and extends through surgical intervention and long-term follow-up. Clinicians must possess the diagnostic skills to recognize the spectrum of AVSD subtypes, accurately interpret echocardiographic findings, and formulate individualized treatment plans. Early identification and timely referral to pediatric cardiology and cardiothoracic surgery are critical for preventing complications such as pulmonary vascular disease. Nurses are crucial in monitoring symptoms, educating patients and their families, and facilitating pre- and postoperative care. Their ability to assess fluid status, recognize signs of heart failure, and provide psychosocial support is essential to patient safety and outcomes.
Effective interprofessional communication and care coordination enhance both patient-centered care and team performance. Pharmacists contribute by ensuring appropriate medication management, particularly in addressing heart failure symptoms and managing anticoagulation if indicated postoperatively. Case managers and social workers help navigate logistical challenges, especially for patients with syndromic conditions who often require additional support services. Regular multidisciplinary team meetings, shared electronic health records, and clearly defined care protocols foster seamless transitions between inpatient and outpatient settings. This collaborative approach improves surgical outcomes, reduces readmissions, and ensures patients and families actively engage in their care journey.
Media
(Click Image to Enlarge)
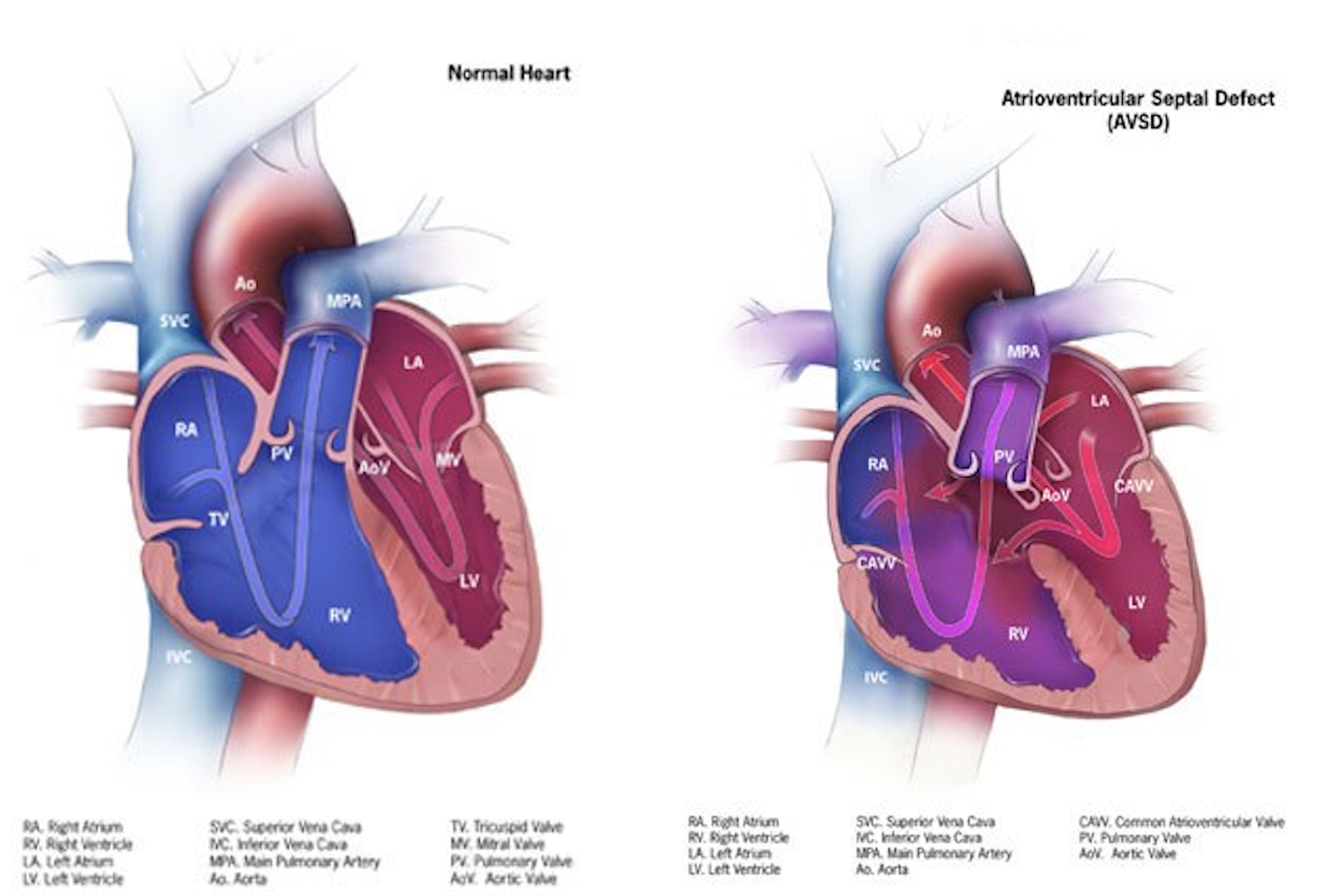
Anatomy of a Heart With Atrioventricular Septal Defects. This illustration compares a normal heart to a heart with atrioventricular septal defects.
Centers for Disease Control and Prevention, Public Domain, via Wikimedia Commons
{kind=link}
(Click Image to Enlarge)
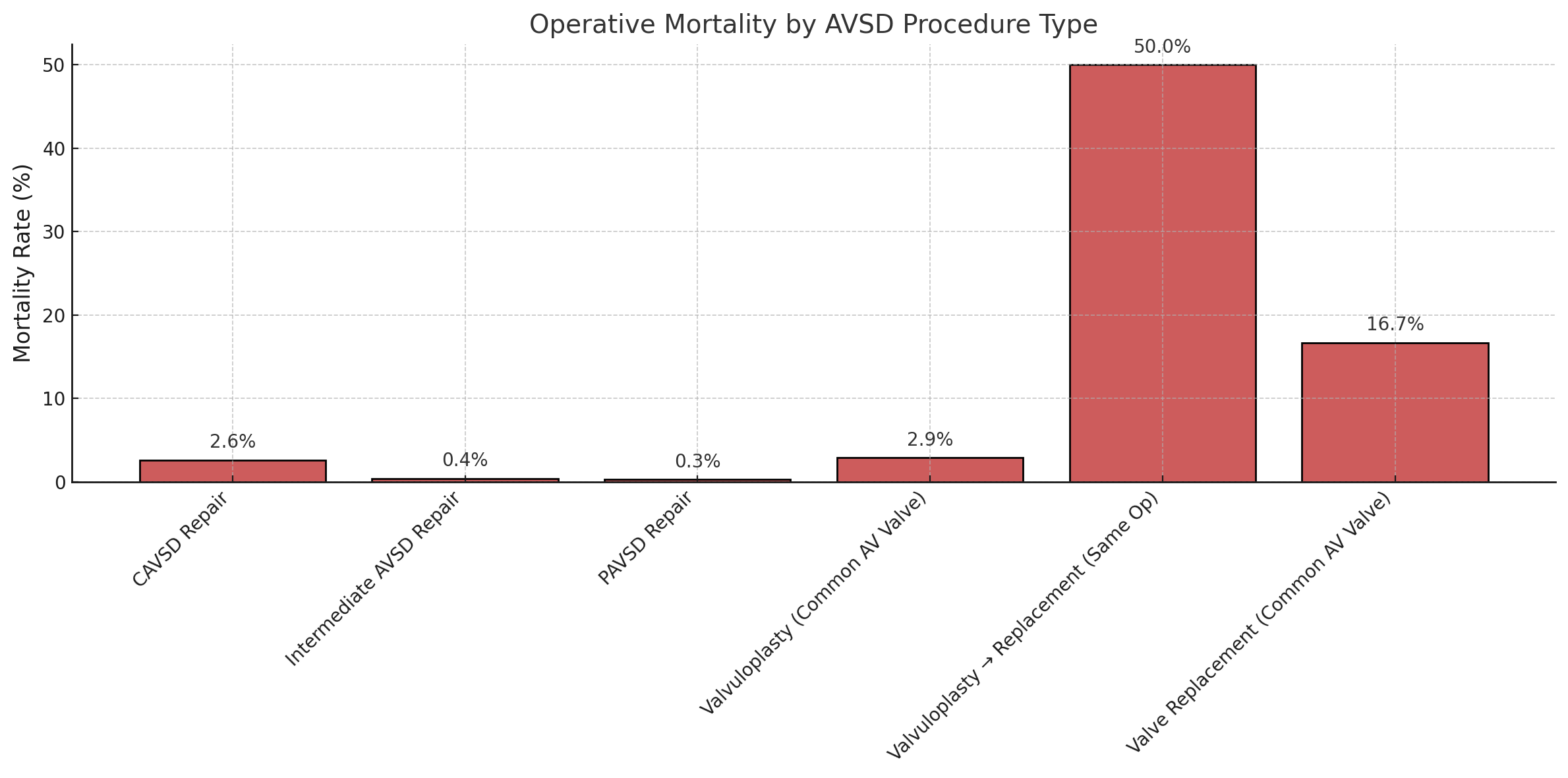
Operative Mortality of Atrioventricular Septal Defect Procedures. This chart shows the operative mortality of procedures to repair atrioventricular septal defect based on data from the Society of Thoracic Surgeons Congenital Heart Surgery Database.
Contributed by MHM Alahmadi, MBBS, MS
(Click Image to Enlarge)
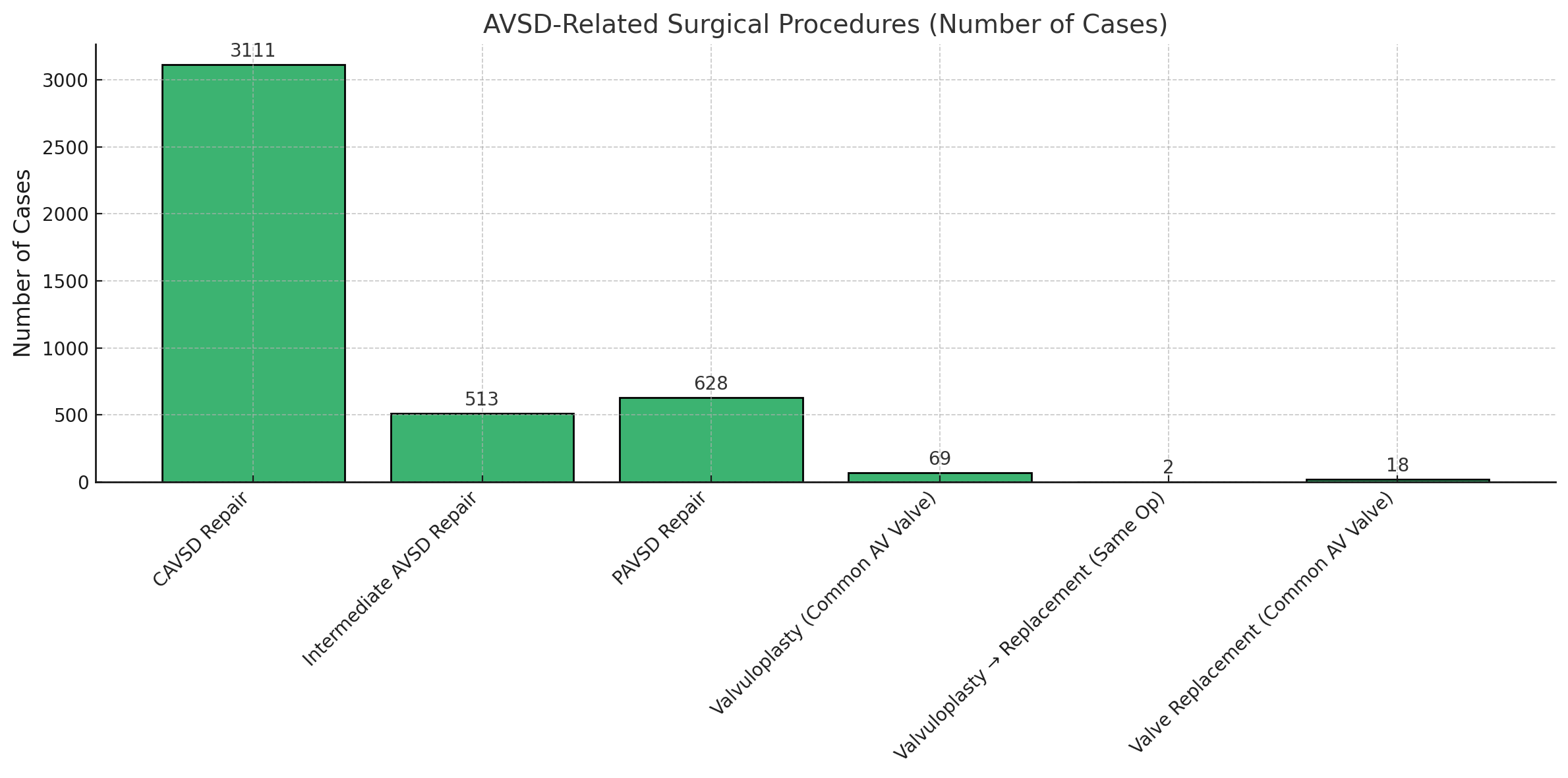
Atrioventricular Septal Defect Procedural Case Numbers. These numbers are based on data from the Society of Thoracic Surgeons Congenital Heart Surgery Database.
Contributed by MHM Alahmadi, MBBS, MS
(Click Image to Enlarge)
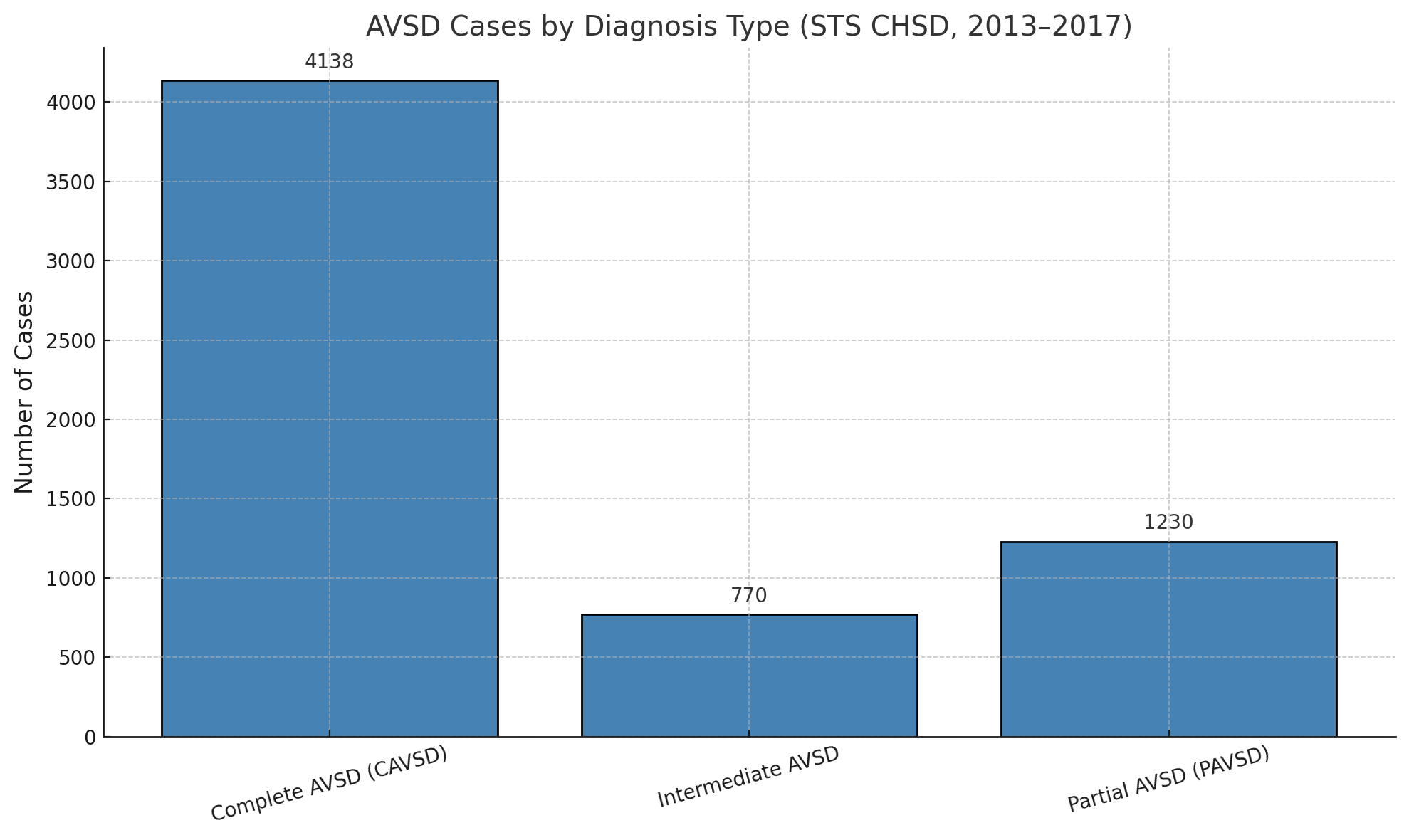
Diagnosed Cases of Atrioventricular Septal Defects. These numbers are based on data from the Society of Thoracic Surgeons Congenital Heart Surgery Database.
Contributed by MHM Alahmadi, MBBS, MS
(Click Image to Enlarge)

Atrioventricular Septal Defect, Rastelli Type A. This shows a Rastelli type A atrioventricular septal defect as viewed from the atrial side.
Contributed by MHM Alahmadi, MBBS, MS
(Click Image to Enlarge)

Atrioventricular Septal Defect, Rastelli Type B. This shows a Rastelli type B atrioventricular septal defect as viewed from the atrial side.
Contributed by MHM Alahmadi, MBBS, MS
(Click Image to Enlarge)

Atrioventricular Septal Defect, Rastelli Type C. This shows a Rastelli type C atrioventricular septal defect as viewed from the atrial side.
Contributed by MHM Alahmadi, MBBS, MS
References
Jacobs JP, Burke RP, Quintessenza JA, Mavroudis C. Congenital Heart Surgery Nomenclature and Database Project: atrioventricular canal defect. The Annals of thoracic surgery. 2000 Apr:69(4 Suppl):S36-43 [PubMed PMID: 10798414]
Calabrò R, Limongelli G. Complete atrioventricular canal. Orphanet journal of rare diseases. 2006 Apr 5:1():8 [PubMed PMID: 16722604]
Tubman TR, Shields MD, Craig BG, Mulholland HC, Nevin NC. Congenital heart disease in Down's syndrome: two year prospective early screening study. BMJ (Clinical research ed.). 1991 Jun 15:302(6790):1425-7 [PubMed PMID: 1829969]
Level 1 (high-level) evidenceBarlow GM, Chen XN, Shi ZY, Lyons GE, Kurnit DM, Celle L, Spinner NB, Zackai E, Pettenati MJ, Van Riper AJ, Vekemans MJ, Mjaatvedt CH, Korenberg JR. Down syndrome congenital heart disease: a narrowed region and a candidate gene. Genetics in medicine : official journal of the American College of Medical Genetics. 2001 Mar-Apr:3(2):91-101 [PubMed PMID: 11280955]
Level 3 (low-level) evidenceAgopian AJ, Moulik M, Gupta-Malhotra M, Marengo LK, Mitchell LE. Descriptive epidemiology of non-syndromic complete atrioventricular canal defects. Paediatric and perinatal epidemiology. 2012 Nov:26(6):515-24. doi: 10.1111/ppe.12006. Epub 2012 Sep 24 [PubMed PMID: 23061687]
Craig B. Atrioventricular septal defect: from fetus to adult. Heart (British Cardiac Society). 2006 Dec:92(12):1879-85 [PubMed PMID: 17105897]
Santoro M, Coi A, Spadoni I, Bianchi F, Pierini A. Sex differences for major congenital heart defects in Down Syndrome: A population based study. European journal of medical genetics. 2018 Sep:61(9):546-550. doi: 10.1016/j.ejmg.2018.05.013. Epub 2018 May 9 [PubMed PMID: 29753092]
Markwald RR, Krook JM, Kitten GT, Runyan RB. Endocardial cushion tissue development: structural analyses on the attachment of extracellular matrix to migrating mesenchymal cell surfaces. Scanning electron microscopy. 1981:(Pt 2):261-74 [PubMed PMID: 7034167]
Level 3 (low-level) evidencePelleri MC, Gennari E, Locatelli C, Piovesan A, Caracausi M, Antonaros F, Rocca A, Donati CM, Conti L, Strippoli P, Seri M, Vitale L, Cocchi G. Genotype-phenotype correlation for congenital heart disease in Down syndrome through analysis of partial trisomy 21 cases. Genomics. 2017 Oct:109(5-6):391-400. doi: 10.1016/j.ygeno.2017.06.004. Epub 2017 Jun 23 [PubMed PMID: 28648597]
Level 3 (low-level) evidenceReeser RS, Salazar AK, Prutton KM, Roede JR, VeDepo MC, Jacot JG. Trisomy 21 Alters Cell Proliferation and Migration of iPSC-Derived Cardiomyocytes on Type VI Collagen. Cellular and molecular bioengineering. 2024 Feb:17(1):25-34. doi: 10.1007/s12195-023-00791-x. Epub 2024 Jan 3 [PubMed PMID: 38435791]
Gittenberger-de Groot AC, Bartram U, Oosthoek PW, Bartelings MM, Hogers B, Poelmann RE, Jongewaard IN, Klewer SE. Collagen type VI expression during cardiac development and in human fetuses with trisomy 21. The anatomical record. Part A, Discoveries in molecular, cellular, and evolutionary biology. 2003 Dec:275(2):1109-16 [PubMed PMID: 14613310]
Trevino CE, Holleman AM, Corbitt H, Maslen CL, Rosser TC, Cutler DJ, Johnston HR, Rambo-Martin BL, Oberoi J, Dooley KJ, Capone GT, Reeves RH, Cordell HJ, Keavney BD, Agopian AJ, Goldmuntz E, Gruber PJ, O'Brien JE Jr, Bittel DC, Wadhwa L, Cua CL, Moskowitz IP, Mulle JG, Epstein MP, Sherman SL, Zwick ME. Identifying genetic factors that contribute to the increased risk of congenital heart defects in infants with Down syndrome. Scientific reports. 2020 Oct 22:10(1):18051. doi: 10.1038/s41598-020-74650-4. Epub 2020 Oct 22 [PubMed PMID: 33093519]
Pugnaloni F, Digilio MC, Putotto C, De Luca E, Marino B, Versacci P. Genetics of atrioventricular canal defects. Italian journal of pediatrics. 2020 May 13:46(1):61. doi: 10.1186/s13052-020-00825-4. Epub 2020 May 13 [PubMed PMID: 32404184]
Chaithra S, Agarwala S, Ramachandra NB. High-risk genes involved in common septal defects of congenital heart disease. Gene. 2022 Oct 5:840():146745. doi: 10.1016/j.gene.2022.146745. Epub 2022 Jul 18 [PubMed PMID: 35863714]
Hong N, Zhang E, Xie H, Jin L, Zhang Q, Lu Y, Chen AF, Yu Y, Zhou B, Chen S, Yu Y, Sun K. The transcription factor Sox7 modulates endocardiac cushion formation contributed to atrioventricular septal defect through Wnt4/Bmp2 signaling. Cell death & disease. 2021 Apr 12:12(4):393. doi: 10.1038/s41419-021-03658-z. Epub 2021 Apr 12 [PubMed PMID: 33846290]
Anderson RH, Ho SY, Falcao S, Daliento L, Rigby ML. The diagnostic features of atrioventricular septal defect with common atrioventricular junction. Cardiology in the young. 1998 Jan:8(1):33-49 [PubMed PMID: 9680269]
Kim JS, Virágh S, Moorman AF, Anderson RH, Lamers WH. Development of the myocardium of the atrioventricular canal and the vestibular spine in the human heart. Circulation research. 2001 Mar 2:88(4):395-402 [PubMed PMID: 11230106]
Rastelli G, Kirklin JW, Titus JL. Anatomic observations on complete form of persistent common atrioventricular canal with special reference to atrioventricular valves. Mayo Clinic proceedings. 1966 May:41(5):296-308 [PubMed PMID: 5932615]
Backer CL, Mavroudis C, Alboliras ET, Zales VR. Repair of complete atrioventricular canal defects: results with the two-patch technique. The Annals of thoracic surgery. 1995 Sep:60(3):530-7 [PubMed PMID: 7677476]
Banka P, Schaetzle B, Komarlu R, Emani S, Geva T, Powell AJ. Cardiovascular magnetic resonance parameters associated with early transplant-free survival in children with small left hearts following conversion from a univentricular to biventricular circulation. Journal of cardiovascular magnetic resonance : official journal of the Society for Cardiovascular Magnetic Resonance. 2014 Oct 7:16(1):73. doi: 10.1186/s12968-014-0073-1. Epub 2014 Oct 7 [PubMed PMID: 25314952]
Berger TJ, Blackstone EH, Kirklin JW, Bargeron LM Jr, Hazelrig JB, Turner ME Jr. Survival and probability of cure without and with operation in complete atrioventricular canal. The Annals of thoracic surgery. 1979 Feb:27(2):104-11 [PubMed PMID: 453968]
ter Heide H, Thomson JD, Wharton GA, Gibbs JL. Poor sensitivity of routine fetal anomaly ultrasound screening for antenatal detection of atrioventricular septal defect. Heart (British Cardiac Society). 2004 Aug:90(8):916-7 [PubMed PMID: 15253968]
Smallhorn JF, Sutherland GR, Anderson RH, Macartney FJ. Cross-sectional echocardiographic assessment of conditions with atrioventricular valve leaflets attached to the atrial septum at the same level. British heart journal. 1982 Oct:48(4):331-41 [PubMed PMID: 7126385]
Level 2 (mid-level) evidenceBacker CL, Stewart RD, Mavroudis C. What is the best technique for repair of complete atrioventricular canal? Seminars in thoracic and cardiovascular surgery. 2007 Fall:19(3):249-57 [PubMed PMID: 17983953]
St Louis JD, Jodhka U, Jacobs JP, He X, Hill KD, Pasquali SK, Jacobs ML. Contemporary outcomes of complete atrioventricular septal defect repair: analysis of the Society of Thoracic Surgeons Congenital Heart Surgery Database. The Journal of thoracic and cardiovascular surgery. 2014 Dec:148(6):2526-31. doi: 10.1016/j.jtcvs.2014.05.095. Epub 2014 Jul 21 [PubMed PMID: 25125206]
Chauhan S. Atrioventricular septal defects. Annals of cardiac anaesthesia. 2018 Jan-Mar:21(1):1-3. doi: 10.4103/aca.ACA_219_17. Epub [PubMed PMID: 29336382]
Overman DM, Baffa JM, Cohen MS, Mertens L, Gremmels DB, Jegatheeswaran A, McCrindle BW, Blackstone EH, Morell VO, Caldarone C, Williams WG, Pizarro C. Unbalanced atrioventricular septal defect: definition and decision making. World journal for pediatric & congenital heart surgery. 2010 Apr:1(1):91-6. doi: 10.1177/2150135110363024. Epub [PubMed PMID: 23804728]
Hoohenkerk GJ, Bruggemans EF, Rijlaarsdam M, Schoof PH, Koolbergen DR, Hazekamp MG. More than 30 years' experience with surgical correction of atrioventricular septal defects. The Annals of thoracic surgery. 2010 Nov:90(5):1554-61. doi: 10.1016/j.athoracsur.2010.06.008. Epub [PubMed PMID: 20971263]
Level 2 (mid-level) evidenceFong LS, Betts K, Bell D, Konstantinov IE, Nicholson IA, Winlaw DS, Orr Y, Australian CAVSD Study Group. Complete atrioventricular septal defect repair in Australia: Results over 25 years. The Journal of thoracic and cardiovascular surgery. 2020 Mar:159(3):1014-1025.e8. doi: 10.1016/j.jtcvs.2019.08.005. Epub 2019 Aug 30 [PubMed PMID: 31590953]
Juaneda I, Pizzulli L, Ferrari P, Molinas R, Diaz J, Azar I, Allub A, Juaneda E, Louis JS, Peirone A, Kathy J. Outcomes of Atrioventricular Septal Defect Repair: Two-Institutions, 10-Year Experience in Cordoba, Argentina. World journal for pediatric & congenital heart surgery. 2025 Jul:16(4):509-515. doi: 10.1177/21501351241305135. Epub 2025 Jan 30 [PubMed PMID: 39885724]
Reynen S, Hövels-Gürich HH, Vazquez-Jimenez JF, Messmer BJ, Sachweh JS. Long-Term Outcome Up To 40 Years after Single Patch Repair of Complete Atrioventricular Septal Defect in Infancy or Childhood. The Thoracic and cardiovascular surgeon. 2021 Dec:69(S 03):e68-e75. doi: 10.1055/s-0041-1740070. Epub 2021 Dec 25 [PubMed PMID: 34953470]
Crawford FA Jr, Stroud MR. Surgical repair of complete atrioventricular septal defect. The Annals of thoracic surgery. 2001 Nov:72(5):1621-8; discussion 1628-9 [PubMed PMID: 11722055]