Introduction
Aplastic anemia is a rare but serious hematologic disorder characterized by the failure of bone marrow to produce enough blood cells, leading to pancytopenia and causing significant morbidity and mortality.[1] This can result in severe complications, including profound fatigue, increased susceptibility to infections, and a heightened risk of hemorrhage.
Aplastic anemia is a life-threatening condition that, if untreated, is associated with very high mortality. However, the advent of bone marrow transplant combined with immunosuppressive therapy (IST) has led to survival rates exceeding 80% to 85%.[2][3] Understanding the underlying causes of aplastic anemia and its pathophysiology is crucial for effective diagnosis and management, as this condition can arise from various factors, including autoimmune disorders, exposure to certain chemicals or medications, and viral infections.[4]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The underlying etiologies of aplastic anemia can be broadly classified into acquired or inherited categories (see Table 1: Aplastic Anemia Etiologies).[5][6][4][1] Acquired aplastic anemia is often linked to environmental factors and external agents, while inherited forms may stem from genetic mutations that affect hematopoiesis.
The most common etiology, idiopathic, accounts for 65%. Fanconi anemia is the most common hereditary cause, presenting in the late first decade with pancytopenia, organ hypoplasia, and bone defects, including abnormal radii, absent thumbs, and short stature. Seronegative hepatitis is responsible for 5% to 10% of total cases. Telomerase defects are found in 5% to 10% of adult-onset aplastic anemia and help in guiding clinical decisions.[7]
Table 1: Aplastic Anemia Etiologies
| Category |
Specific Agents |
| Idiopathic |
|
| Drug-Induced
|
|
| Chemical and Environmental Toxins |
|
| Radiation and Cancer Therapy |
|
| Infectious Causes |
|
| Autoimmune and Immune-mediated Disorders |
|
| Associated Hematologic Disorders |
|
| Hereditary Syndromes (in children/young adults) |
|
| Endocrine and Nutritional Factors |
|
| Tumors and Thymic Abnormalities |
|
Epidemiology
Aplastic anemia is a rare disorder with a bimodal age distribution. Research indicates that the incidence ranges from 0.6 to 6.1 cases for every million individuals; this rate is primarily derived from analyses of historical death registries. The ratio of males to females is roughly 1:1. Younger age, male predominance, and higher consanguinity suggest that genetic factors may play a role in the etiology of aplastic anemia among the South Asian population.[1]
The prevalence of adult aplastic anemia in Thailand and Taiwan exceeds that found in Western nations, with a particularly high incidence among older individuals.[8][9] Aplastic anemia can affect individuals across all age groups, but the initial peak in incidence occurs within the first 30 years, followed by a secondary peak in older patients (those older than 60).[10] Patients with severe forms of the disease experience lower 5-year survival rates.[11] An increased incidence of aplastic anemia has been associated with the inheritance of certain human leukocyte antigen (HLA) class I alleles that vary among populations.[12]
A higher prevalence of autoimmune anemia has been associated with the genetic transmission of specific HLA class I alleles that exhibit differences across different populations.[12]
Pathophysiology
Aplastic anemia is characterized by the failure of hematopoietic stem cells in the bone marrow, leading to pancytopenia. The pathogenesis involves both immune-mediated and intrinsic mechanisms, as well as genetic and environmental factors. This multifactorial pathophysiology highlights the complexity of aplastic anemia and underscores the importance of a targeted therapeutic approach.
Immune-Mediated Suppression
The following pathophysiological processes are associated with autoimmune responses, resulting in the depletion of hematopoietic stem cells in aplastic anemia:
-
T-cell-mediated cytotoxicity: Autoreactive T-helper lymphocytes (T1) secrete cytokines (eg, IFN-γ and TNF), thus triggering the apoptotic pathway in hematopoietic stem cells. As a result, this observation supports the theory that immunosuppressive treatments targeting T-cells (including antithymocyte globulin) have demonstrated effectiveness for numerous patients.[13][14]
-
HLA class I alleles: Certain HLA class I alleles have been identified as either elevated or reduced in cases of aplastic anemia, suggesting that CD8+ T cells might be targeting the bone marrow as a result of immune reactions similar to those seen in viral infections or autoimmune responses.[15][12][16][17]
-
Hepatitis-associated aplastic anemia (HAAA): In cases of HAAA, severe autoimmune hepatitis typically presents before aplastic anemia, frequently arising within a 6-month timeframe.[13] HAAA exhibits a distinct immune signature marked by oligoclonality and a diminished count of CD4+ T cells compared to idiopathic aplastic anemia.[18]
-
Cytokine dysregulation: Pro-inflammatory cytokines, eg, IFN-γ and TNF-α, exert an inhibitory effect on hematopoiesis without inducing direct harm to hematopoietic stem cells.[19]
-
Myeloid dendritic cell: The levels of mDC1 are higher in cases of severe aplastic anemia and are associated with the intensity of the disease. This indicates that mDC1 could play a crucial role and serve as a potential therapeutic target in SAA.[20]
Intrinsic Stem Cell Defects
Stem cell abnormalities
Stem cell abnormalities contribute to bone marrow insufficiency by impairing the function of hematopoietic stem cells, which lose their ability to replicate or mature effectively. This failure in normal hematopoiesis can lead to the development of severe hematologic conditions, including myelodysplastic syndrome.[13]
Telomere impairment
Telomere impairment also plays a crucial role in the depletion of hematopoietic stem cells. Shortened telomeres impact stem cell longevity and function, thereby affecting the effectiveness of various treatment strategies. A clear understanding of telomere dynamics informs clinical decision-making and contributes to improved survival outcomes in patients undergoing stem cell transplantation.[7][21]
External Triggers
Several external factors can contribute to the development of aplastic anemia. Exposure to specific medications, toxic chemicals, and ionizing radiation can directly damage hematopoietic stem cells, impairing their ability to produce adequate blood cells.[22][23] Agents, eg, chemotherapy drugs, benzene, and other industrial solvents have been associated with this form of marrow suppression.
Infectious agents also play a significant role in triggering aplastic anemia. Certain viral infections, including Epstein-Barr virus (EBV), can activate cytotoxic T-cells, which may lead to immune-mediated destruction of bone marrow precursors.[24][25] Additionally, COVID-19 has emerged as a contributing factor, with reports linking the virus to cases of aplastic anemia through similar immune dysregulation mechanisms.
Clonal Evolution
Aplastic anemia may progress to paroxysmal nocturnal hemoglobinuria (PNH), myelodysplastic syndromes (MDS), or acute myeloid leukemia (AML) due to genetic alterations and chromosomal irregularities.[26][27][28][29][30]
Genetic and Environmental Factors
Specific genetic variations may predispose certain individuals to heightened susceptibility to hematopoietic stem cell depletion, thereby increasing the probability of developing aplastic anemia.[12]
History and Physical
Clinical Evaluation
Patients with aplastic anemia typically present with symptoms of pancytopenia, including fatigue and dyspnea (anemia), infections and fever (neutropenia), and mucosal bleeding or petechiae (thrombocytopenia). Infections are often bacterial; severe neutropenia increases the risk of invasive fungal infections. Some cases are asymptomatic and are detected via abnormal laboratory blood counts, including complete blood count (CBC), reticulocyte count, bone marrow biopsy, and testing for inherited marrow failure syndromes.
The urgency of therapeutic intervention depends on the severity of cytopenia and the patient's clinical condition; hence, it requires immediate consultation with both a hospitalist and a hematologist/oncologist, and may necessitate inpatient care.[31][32]
Clinical History
A thorough history and physical examination are essential in assessing the patient's overall health and identifying potential causes of aplastic anemia. A detailed review of the medical history, including any previous exposure to toxic substances or medications, family history of blood disorders, and current symptom presentation, is vital in guiding further diagnostic tests and treatment plans.
Key historical points that should be assessed include:
- Recent exposure to drugs, toxins, or radiation
- Recent viral illnesses (eg, hepatitis, EBV, parvovirus B19, COVID-19)
- Family history of inherited marrow failure syndromes
- Cytopenia-related symptoms: infections, bleeding, fatigue
Physical Examination
The physical examination may reveal clinical signs, including pallor, jaundice, or bleeding, which can be crucial in evaluating the severity of the condition and determining the need for prompt medical attention. Other findings associated with aplastic anemia include:
-
Signs of pancytopenia
-
Anemia: Pallor, tachycardia, dyspnea
-
Neutropenia: Fever, signs of infection
-
Thrombocytopenia: Petechiae, bruising, mucosal bleeding
-
-
Absence of liver, spleen, and lymph node enlargement; notable enlargement of these structures may suggest another diagnosis
-
Inherited aplastic anemia syndromes (eg, Fanconi anemia, dyskeratosis congenita, Diamond-Blackfan anemia, Schwachman-Diamond syndrome), including short stature, abnormal thumbs, cardiac defects, abnormal skin pigmentation, nail dystrophy, and other congenital abnormalities [33]
Evaluation
Diagnostic Evaluation
Laboratory evaluations and a bone marrow biopsy are crucial for confirming the diagnosis and evaluating the severity of cytopenia, which consists of a CBC, reticulocyte count, and peripheral blood smear.[33][34] These diagnostic methods enable the distinction between the various causes of cytopenia and aid in developing suitable treatment strategies based on the identified underlying condition. The following findings on diagnostic studies help in excluding differential diagnoses or identifying aplastic anemia:
- Peripheral blood findings
- CBC with differential: Shows pancytopenia
- Reticulocyte count: Low
- Telomere length/chromosome breakage test: Rule out inherited marrow failure syndromes
- Flow cytometry: Aids in evaluating for MDS, leukemia, or PNH by identifying and distinguishing hypoplastic MDS from aplastic anemia.[35]
- Bone marrow
- Biopsy:
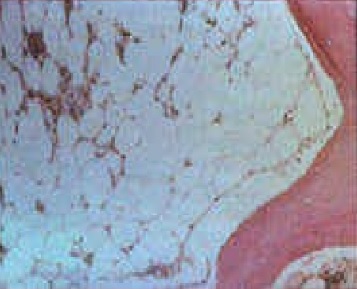
- Hypocellular marrow with fatty replacement (see Image. Aplastic Anemia Bone Marrow)
- No fibrosis or malignant infiltration
- Aspirate: Often results in a “dry tap”
- Biopsy:
-
FISH/SNP arrays: Helpful when the aspirate provides a limited number of cells.[37]
-
Next generation sequencing (NGS) panel: Could uncover mutations (eg, DNMT3A, ASXL1) [38][39]
Diagnostic Criteria for Aplasitc Anemia
Diagnosis of aplastic anemia typically requires a combination of clinical findings, laboratory tests, and the exclusion of other hematological disorders.[33][40]
Aplastic anemia is a clinical-pathologic diagnosis that requires both of the following:
-
Bone marrow hypoplasia/aplasia without an infiltrative process
-
The presence of ≥2 of the following cytopenia findings:
-
Reticulocytes <40,000/µL (or <1%)
-
ANC <500/µL
-
Platelets <20,000/µL
-
Severity Grading
Aplastic anemia can be categorized according to the severity of bone marrow insufficiency and the presence of peripheral blood cytopenia, thereby guiding clinical management strategies and prognostic assessments (Table 2: Severity Grading).[40]
Table 2: Severity Grading
| Grade | Bone Marrow | Peripheral Blood |
| Severe Aplastic Anemia (SAA) | Cellularity <25%, or 25% to 50% with <30% hematopoietic cells | ≥2 of the following: -absolute neutrophil count <500/µL - Platelets <20,000/µL - Reticulocytes <60,000/µL (some use <40,000/µL) |
| Very Severe Aplastic Anemia (vSAA) | Meets SAA criteria |
absolute neutrophil count <200/µL |
| Nonsevere Aplastic Anemia | Same marrow findings as SAA | Cytopenia’s not meeting SAA or vSAA thresholds |
Treatment / Management
Management of Aplastic Anemia in Adults
The treatment of aplastic anemia is highly dependent on the severity of the condition. A comprehensive pretreatment evaluation is crucial to determine the most effective and personalized treatment strategy for individuals affected by this complex and potentially life-threatening condition.
Management of aplastic anemia focuses on addressing the underlying cause, emphasizing the importance of an accurate diagnosis and characterization of the anemia. If the underlying cause is identified as exposure to a drug or cytotoxic chemical, discontinuation of the offending agent is recommended, if possible. Aplastic anemia associated with pregnancy is typically self-limiting and resolves with delivery, but relapses are common.[41] In cases involving thymoma, patients often experience full recovery of bone marrow function following thymectomy.[42][43][44](B3)
Pretreatment evaluation
In cases where no reversible etiology is identified, the therapeutic approach is contingent upon factors, eg, the patient's age, disease severity, donor availability, and the patient's overall functional status.
The initial evaluation of aplastic anemia must exclude other causes of pancytopenia, as significant differences in treatment approaches exist. This includes a thorough history, physical examination, laboratory tests, bone marrow examination, cytogenetics, and molecular studies.
Exclusion of differential diagnoses
Inherited bone marrow failure syndromes (IBMFS) should be considered, particularly in patients younger than 40 years of age.[45] Chromosomal breakage analysis and telomere length analysis are recommended for these patients to identify any underlying genetic abnormalities that may influence treatment decisions and prognosis. Additionally, NGS panels should be obtained to evaluate for genetic mutations causing bone marrow failure, as this impacts factors related to hematopoietic stem cell transplantation (HSCT).
Severe Aplastic Anemia and Very Severe Aplastic Anemia
Patients with severe aplastic anemia (SAA) or very severe aplastic anemia (vSAA) require prompt treatment to reduce the risk of life-threatening infections and limit complications from ongoing transfusions and medications. A short-term delay in initiating therapy is acceptable for treating serious bacterial infections or sepsis, but not for fungal infections unless they can be managed through surgical means.
Medically fit patients
For patients aged 40 or younger who are diagnosed with severe aplastic anemia (SAA) or very severe aplastic anemia (vSAA) and possess a rapidly accessible matched related donor (MRD), the utilization of allogeneic HSCT is favored in comparison to immunosuppressive therapy (IST).[46][47][48][49][50][51] In instances where an MRD is not available, the initiation of IST is warranted.(A1)
For patients older than 40, triple IST (horse antithymocyte globulin [hATG], cyclosporine, and eltrombopag) is advocated due to its superior clinical outcomes compared to IST without eltrombopag.[52][53][54][55][56][57][58] Recent investigations indicate that the incorporation of Avatrombopag (AVA) into IST enhances both the response rate and the quality of response in patients diagnosed with SAA, whilst maintaining a favorable safety profile.[59](B2)
Medically unfit or frail patients
For individuals who are not deemed suitable candidates for intensive IST or HSCT, therapeutic interventions are tailored to alleviate symptoms and enhance overall quality of life while minimizing the occurrence of adverse effects. IST should be regarded as the primary treatment modality for the majority of older patients diagnosed with SAA.[60]
Moderate Aplastic Anemia
Indications for the need for therapeutic intervention for moderate aplastic anemia (MAA) are primarily determined by the dependence on red blood cell (RBC) transfusions, which serves as the most frequent reason for initiating treatment, usually becoming apparent after administering 6 to 10 units of RBCs or when the patient's quality of life is significantly affected due to cytopenias. In addition, the ongoing presence of severe neutropenia or thrombocytopenia may also necessitate medical intervention.
For patients considered medically stable, the initial treatment strategy may include lower-intensity IST or monotherapy with eltrombopag. Intensive immunosuppressive therapy is considered suitable for specific individuals, while HSCT is rarely viewed as a first-line option but may be chosen by younger patients facing significant transfusion needs or who have coexisting infections. In vulnerable patients, supportive care is prioritized, focusing on symptom relief and improving quality of life.
Treatment Approach for Severe Aplastic Anemia
Various management strategies may be used in patients with SAA, including pharmacologic treatment, supportive therapies, and surveillance.
Hematopoietic stem cell transplantation (HSCT)
Allogeneic HSCT can induce remission in aplastic anemia. HSCT fulfills the dual role of restoring the pool of hematopoietic stem/progenitor cells while simultaneously replacing the immune system, which is involved in their loss. The absolute neutrophil count before allogeneic HSCT has been recognized as a crucial prognostic factor in adult populations diagnosed with aplastic anemia.[61]
Triple immunosuppressive therapy (IST)
Horse antithymocyte globulin (hATG) functions by eliminating antigen-specific T lymphocytes and facilitating hematologic responses in cases of aplastic anemia. Cyclosporine inhibits the synthesis and secretion of interleukin-2 (IL-2) while concurrently blocking the IL-2-mediated activation of quiescent T lymphocytes. Eltrombopag, classified as a non-peptide agonist of thrombopoietin, augments platelet counts and stimulates intracellular signal transduction pathways that promote the proliferation and differentiation of hematopoietic progenitor cells within the marrow.[54]
The implementation of triple IST is favored over the combination of hATG and cyclosporine alone due to the superior clinical outcomes observed.[53][54][62]
Lower-intensity aplastic anemia treatments
Eltrombopag is a non-peptide thrombopoietin receptor (TPO-R) agonists that serve to enhance the proliferation of hematopoietic and megakaryocyte progenitor cells.[63] Administering this medication either in a fasted state or concurrently with a meal that is low in calcium content is recommended.(B3)
Reduced-intensity immunosuppressive therapy (IST), which involves the use of cyclosporine A either alone or in combination with alternative immunosuppressive agents, has yielded suboptimal therapeutic outcomes when compared to the standard regimen of horse antithymocyte globulin (hATG) combined with cyclosporine. In patients with SAA who present with compromised cardiac or renal function, the preferred clinical approach focuses on optimizing underlying medical conditions to enable the use of full triple IST, rather than defaulting to a lower-intensity regimen.
Cyclosporine monotherapy in MAA has demonstrated a response rate of 46%, a transfusion-independence rate of 67%, and an overall survival rate of 93% at 180 days. The combination of cyclosporine and eltrombopag, excluding hATG, remains under investigation for older or medically frail patients diagnosed with MAA. Cyclosporine used in conjunction with levamisole has achieved a 100% response rate in newly diagnosed MAA cases and an 87% response rate in chronic MAA cases.[33]
Monitoring
Patients administered hATG are susceptible to experiencing infusion reactions and serum sickness. Monitoring for these adverse effects is crucial, and premedication with antihistamines or corticosteroids might be warranted to alleviate reactions throughout the therapeutic regimen.[64][65][66] Consistent evaluation of hematological parameters, renal function, and trough concentrations of cyclosporine is essential.
Clinicians should monitor for secondary hemochromatosis and administer iron chelation therapy when clinically necessary. The use of growth factors, eg, erythropoietin or granulocyte colony-stimulating factors, is not recommended due to a lack of precursor cells required to achieve sufficient physiological responses.
Supportive therapy
Transfusion support plays a crucial role in managing patients with severe anemia or thrombocytopenia resulting from aplastic anemia. RBC transfusions help alleviate symptoms such as fatigue and dyspnea, while platelet transfusions reduce the risk of spontaneous bleeding and help maintain hemostasis. These interventions contribute significantly to improving quality of life and overall clinical stability.
Preventing infections remains a top priority, as infections represent the most common cause of mortality in individuals with aplastic anemia. The development of fever in the context of an absolute neutrophil count below 500/µL constitutes a medical emergency requiring urgent evaluation and intervention. Prophylactic measures against Pneumocystis jirovecii should include agents such as sulfamethoxazole/trimethoprim or pentamidine. Antifungal and antiviral prophylaxis should also be tailored to the patient's specific risk profile to reduce the likelihood of life-threatening infections.[67]
Follow-up and treatment responses
Patients undergoing IST for aplastic anemia require consistent monitoring to assess treatment response, detect toxicities, and identify signs of clonal progression.
During the first 6 months of treatment, outpatient visits occur every 2 to 4 weeks. Biweekly evaluations include CBC, reticulocyte counts, liver transaminases, and cyclosporine A levels. Bone marrow examinations are conducted at the 3-month and 6-month marks, especially for patients showing partial responses, to evaluate marrow cellularity and treatment effectiveness.
After the initial 6-month period, monitoring continues for patients who maintain stability on eltrombopag and/or cyclosporine A. Once therapy is discontinued, follow-up visits are scheduled every 3 to 6 months, gradually extending to annual evaluations after 2 to 3 years, depending on the patient's clinical course.
Assessment of hematologic response involves several benchmarks. Transfusion independence and improved peripheral blood counts indicate a favorable outcome. A complete response includes transfusion independence, hemoglobin levels above 10 g/dL, and platelet counts exceeding 50,000/µL. A partial response indicates improved blood counts or decreased transfusion requirements, but does not meet the full response criteria. Refractory disease shows minimal or no improvement despite therapy, while relapse is defined by a subsequent decline in blood counts after an initial hematologic response.
Refractory Aplastic Anemia
Refractory aplastic anemia is a challenging condition that often requires alternative therapeutic strategies, including the consideration of IST or HSCT for eligible patients.
HSCT is the preferred method for eligible patients, particularly when a matched related donor is available. For patients who do not qualify for HCT, options include Eltrombopag or hATG-based treatments.[68]
Relapsing Aplastic Anemia
Relapse occurs in up to one-third of patients with SAA. Management is guided by serial blood counts and bone marrow exams. A second course of IST can be effective in 55% to 60% of patients, but salvage HSCT may be a more preferable treatment option for relapses or refractory cases.
Differential Diagnosis
The differential diagnosis of pancytopenia encompasses several conditions that may resemble aplastic anemia. Myelophthisic syndrome, characterized by the replacement of normal bone marrow by tumors or fibrosis, is caused by solid tumor metastases (eg, lung, breast, prostate), myeloid or lymphoid malignancies, or disorders like Gaucher disease. Unlike aplastic anemia, these conditions typically present with marrow that is not hypocellular, reflecting the underlying disease.
Isolated hematopoietic lineage failure, eg, agranulocytosis or pure red cell aplasia, shares etiologies with aplastic anemia but presents with symptoms specific to the affected cell line.
Reversible marrow suppression, caused by chemotherapy, radiation, sepsis, or viral infections, can mimic aplastic anemia but is distinguished by the transient nature of blood count abnormalities and the absence of hypocellularity on bone marrow biopsy.[32]
Additionally, megaloblastic anemia, hypersplenism, malignancies, myelofibrosis, infections, and inherited bone marrow failure syndromes (eg, Fanconi anemia, Shwachman-Diamond syndrome) must be considered.[69] These conditions have distinguishing features, including hypersegmented neutrophils and macroovalocytes in megaloblastic anemia, or splenomegaly in hypersplenism.[70][20]
Prognosis
Over the past 3 decades, the prognosis for patients with aplastic anemia has improved significantly due to advances in IST and HSCT, with a 10-year survival rate increased, especially for sibling-matched HCT.[71][72] However, IST carries risks of relapse and late clonal evolution, with leukocyte telomere length linked to poor survival and increased relapse.[7][73]
In severe aplastic anemia (SAA), survival rates range from 80% to 90%, with younger patients and those with higher reticulocyte and lymphocyte counts experiencing better outcomes.[74] The presence of preexisting anti-HLA antibodies negatively affects platelet recovery and overall prognosis in SAA patients undergoing allogeneic HCT.[75] Despite the benefits of HSCT, complications, eg, graft-versus-host disease and graft failure, remain significant risks.[76]
Factors like higher ECOG scores and infections are associated with poorer posttransplant outcomes.[76] Delayed HLA-PB haplo-HSCT for severe aplastic anemia yielded poor outcomes. Rapid referral for HSCT is critically required.[77] Haplo-HSCT should be considered an effective alternative for patients with SAA without a matched sibling donor.[78]
Complications
The most common complications of aplastic anemia include bleeding, infections, or transformation to lymphoproliferative disorders. These conditions are managed through surveillance and symptomatic treatment, which may include antibiotics, chemotherapy, and transfusions.
Deterrence and Patient Education
Deterrence in aplastic anemia primarily involves minimizing exposure to known risk factors and addressing modifiable contributors to bone marrow failure. Patients should receive guidance on avoiding toxic chemicals, eg, benzene, certain pesticides, and medications known to affect hematopoiesis. Individuals with a family history of inherited marrow failure syndromes may benefit from genetic counseling to assess potential risks and guide early screening. Prompt evaluation and management of viral infections, including hepatitis viruses and Epstein-Barr virus, can also reduce the likelihood of immune-mediated marrow suppression. In cases involving drug-induced aplastic anemia, immediate discontinuation of the offending agent is essential to prevent progression.
Patient education plays a crucial role in enhancing outcomes and fostering self-management. Clinicians should inform patients about the signs and symptoms of cytopenia, eg, fatigue, infections, and bleeding, and emphasize the importance of routine follow-up visits, laboratory monitoring, and adherence to medication. Education on infection prevention strategies, including hand hygiene, avoidance of crowded places during periods of neutropenia, and prompt reporting of fever, remains crucial. Patients should also understand the rationale behind treatment choices, including immunosuppressive therapy, hematopoietic stem cell transplantation, and supportive care, so they can actively participate in shared decision-making and recognize the need for urgent care in cases of relapse or complications.
Pearls and Other Issues
Pancytopenia, defined as the simultaneous reduction of erythrocytes, leukocytes, and platelets, is a hematologic finding with a broad differential diagnosis that includes bone marrow failure syndromes, infiltrative diseases, nutritional deficiencies, and hematologic malignancies. Aplastic anemia represents a distinct etiology of pancytopenia, characterized by immune-mediated destruction or suppression of hematopoietic stem cells, resulting in a markedly hypocellular bone marrow without malignant infiltration or fibrosis.
This condition must be differentiated from other causes of pancytopenia, eg, myelophthisic syndrome, which results from the pathological replacement of normal hematopoietic marrow with abnormal tissue. Etiologies of myelophthisic syndrome include metastatic solid tumors (eg, breast, lung, or prostate cancer), hematologic malignancies (eg, acute myeloid leukemia), myelofibrosis, hemophagocytic lymphohistiocytosis, osteopetrosis, and lysosomal storage disorders such as Gaucher disease. In these cases, bone marrow biopsy typically reveals preserved or increased cellularity with evidence of infiltrative or fibrotic processes, rather than the hypocellularity observed in aplastic anemia.
Additionally, isolated failure of a single hematopoietic lineage may occur, manifesting as agranulocytosis or pure red cell aplasia, and may share etiologic factors with aplastic anemia, including drug exposures (eg, propylthiouracil) and paraneoplastic syndromes (eg, thymoma-associated red cell aplasia). Unlike aplastic anemia, these conditions are associated with cytopenia limited to a specific cell line, and clinical manifestations correspond to the deficient lineage rather than global bone marrow suppression. Accurate diagnosis relies on a comprehensive evaluation including peripheral blood counts, bone marrow biopsy, and assessment for underlying systemic or malignant processes, as treatment strategies vary significantly depending on the etiology.
Enhancing Healthcare Team Outcomes
The successful management of aplastic anemia relies on a highly coordinated interprofessional approach that emphasizes patient-centered care, clinical vigilance, and evidence-based decision-making. Physicians and advanced practitioners, including hematologists and transplant specialists, lead diagnostic and therapeutic planning by assessing disease severity and determining the appropriateness of immunosuppressive therapy or hematopoietic stem cell transplantation. Close collaboration with infectious disease consultants ensures early identification and treatment of infections, particularly in the setting of profound neutropenia. Nurses play a central role in daily patient care, administering medications, educating patients and families, and monitoring for signs of bleeding, infection, or drug toxicity. Pharmacists contribute by reviewing complex treatment regimens, adjusting dosages based on renal or hepatic function, and ensuring safe administration of immunosuppressants and prophylactic antimicrobials. Coordination with hospitalists ensures seamless transitions across care settings, while dietitians provide tailored nutritional counseling, especially for patients following neutropenic dietary precautions.
Effective interprofessional communication and structured care pathways remain vital for enhancing safety and optimizing outcomes. Regular case conferences and handoffs among team members promote early recognition of complications and reinforce shared decision-making that aligns with patient goals. Nurses and advanced practitioners serve as essential patient advocates, reinforcing education on infection prevention, dietary restrictions, and activity limitations to prevent trauma-related hemorrhage. Nutritional counseling plays a critical role; neutropenic dietary precautions should be implemented, with avoidance of high-risk foods such as unpasteurized dairy products, raw or undercooked meats, and unwashed fruits and vegetables, which may harbor pathogenic microorganisms. Hormonal therapy may be discussed by specialists in collaboration with gynecologists for the management of menorrhagia in premenopausal women.
Comprehensive discharge planning and outpatient follow-up require consistent communication across disciplines to ensure continuity of care, medication adherence, and long-term monitoring. Through deliberate teamwork and clear role delineation, the interprofessional team enhances treatment success, minimizes risks, and supports patients as they navigate the complexities of aplastic anemia care.[71][72]
Media
(Click Image to Enlarge)
Aplastic Anemia Bone Marrow. Image demonstrating histologic findings of bone marrow biopsy in a patient with aplastic anemia.
Contributed by Ruozhi Xiao
References
Ahmed P, Chaudhry QUN, Satti TM, Mahmood SK, Ghafoor T, Shahbaz N, Khan MA, Satti HS, Akram Z, Iftikhar R. Epidemiology of aplastic anemia: a study of 1324 cases. Hematology (Amsterdam, Netherlands). 2020 Dec:25(1):48-54. doi: 10.1080/16078454.2019.1711344. Epub [PubMed PMID: 31906834]
Level 3 (low-level) evidenceScheinberg P. Progress in medical therapy in aplastic anemia: why it took so long? International journal of hematology. 2024 Mar:119(3):248-254. doi: 10.1007/s12185-024-03713-3. Epub 2024 Feb 26 [PubMed PMID: 38403842]
Boddu P, Garcia-Manero G, Ravandi F, Borthakur G, Jabbour E, DiNardo C, Jain N, Daver N, Pemmaraju N, Anderlini P, Parmar S, Kc D, Akosile M, Pierce SA, Champlin R, Cortes J, Kantarjian H, Kadia T. Clinical outcomes in adult patients with aplastic anemia: A single institution experience. American journal of hematology. 2017 Dec:92(12):1295-1302. doi: 10.1002/ajh.24897. Epub 2017 Sep 25 [PubMed PMID: 28850699]
Level 2 (mid-level) evidenceShallis RM, Ahmad R, Zeidan AM. Aplastic anemia: Etiology, molecular pathogenesis, and emerging concepts. European journal of haematology. 2018 Dec:101(6):711-720. doi: 10.1111/ejh.13153. Epub 2018 Oct 10 [PubMed PMID: 30055055]
Steensma DP. Revisiting the first reported case of aplastic anaemia. British journal of haematology. 2024 Feb:204(2):455-458. doi: 10.1111/bjh.19241. Epub 2023 Dec 3 [PubMed PMID: 38044033]
Level 3 (low-level) evidenceJang T, Burnside RD, Chaffin J, Seifert R, Hiemenz JW. Severe aplastic anemia with acquired X chromosome clonality as a sole abnormality. Annals of hematology. 2025 Jan:104(1):815-819. doi: 10.1007/s00277-024-06166-0. Epub 2024 Dec 31 [PubMed PMID: 39738592]
Strauss JD, Brown DW, Zhou W, Dagnall C, Yuan JM, Im A, Savage SA, Wang Y, Rafati M, Spellman SR, Gadalla SM. Telomere length and clonal chromosomal alterations in peripheral blood of patients with severe aplastic anaemia. British journal of haematology. 2024 Sep:205(3):1180-1187. doi: 10.1111/bjh.19681. Epub 2024 Aug 5 [PubMed PMID: 39103182]
Norasetthada L, Wongkhantee S, Chaipokam J, Charoenprasert K, Chuncharunee S, Rojnuckarin P, Sirijerachai C, Wanachiwanawin W, Issaragrisil S, Thai Aplastic Anemia Study Group. Adult aplastic anemia in Thailand: incidence and treatment outcome from a prospective nationwide population-based study. Annals of hematology. 2021 Oct:100(10):2443-2452. doi: 10.1007/s00277-021-04566-0. Epub 2021 Jul 16 [PubMed PMID: 34269837]
Li SS, Hsu YT, Chang C, Lee SC, Yen CC, Cheng CN, Chen JS, Lin SH, Chang KC, Chen TY. Incidence and treatment outcome of aplastic anemia in Taiwan-real-world data from single-institute experience and a nationwide population-based database. Annals of hematology. 2019 Jan:98(1):29-39. doi: 10.1007/s00277-018-3486-3. Epub 2018 Sep 3 [PubMed PMID: 30178191]
Young NS, Kaufman DW. The epidemiology of acquired aplastic anemia. Haematologica. 2008 Apr:93(4):489-92. doi: 10.3324/haematol.12855. Epub [PubMed PMID: 18379007]
Lee GWC, Yeap MY, Tan XY, Tang ASO, Ho YF, Law KB, Lee SWF, Chew LP, Wong LLL. A multicentre, retrospective study of epidemiology and outcome of aplastic anaemia among adult population in Sabah and Sarawak from year 2006 to 2017. The Medical journal of Malaysia. 2024 Nov:79(6):749-756 [PubMed PMID: 39614794]
Level 2 (mid-level) evidenceOlson TS, Frost BF, Duke JL, Dribus M, Xie HM, Prudowsky ZD, Furutani E, Gudera J, Shah YB, Ferriola D, Dinou A, Pagkrati I, Kim S, Xu Y, He M, Zheng S, Nijim S, Lin P, Xu C, Nakano TA, Oved JH, Carreno BM, Bolon YT, Gadalla SM, Marsh SG, Paczesny S, Lee SJ, Monos DS, Shimamura A, Bertuch AA, Gragert L, Spellman SR, Babushok DV. Pathogenicity and impact of HLA class I alleles in aplastic anemia patients of different ethnicities. JCI insight. 2022 Nov 22:7(22):. doi: 10.1172/jci.insight.163040. Epub 2022 Nov 22 [PubMed PMID: 36219480]
Giudice V, Selleri C. Aplastic anemia: Pathophysiology. Seminars in hematology. 2022 Jan:59(1):13-20. doi: 10.1053/j.seminhematol.2021.12.002. Epub 2022 Jan 5 [PubMed PMID: 35491054]
Young NS. Aplastic Anemia. The New England journal of medicine. 2018 Oct 25:379(17):1643-1656. doi: 10.1056/NEJMra1413485. Epub [PubMed PMID: 30354958]
Zaimoku Y, Patel BA, Adams SD, Shalhoub R, Groarke EM, Lee AAC, Kajigaya S, Feng X, Rios OJ, Eager H, Alemu L, Quinones Raffo D, Wu CO, Flegel WA, Young NS. HLA associations, somatic loss of HLA expression, and clinical outcomes in immune aplastic anemia. Blood. 2021 Dec 30:138(26):2799-2809. doi: 10.1182/blood.2021012895. Epub [PubMed PMID: 34724566]
Level 2 (mid-level) evidenceBen Hamza A, Welters C, Stadler S, Brüggemann M, Dietze K, Brauns O, Brümmendorf TH, Winkler T, Bullinger L, Blankenstein T, Rosenberger L, Leisegang M, Kammertöns T, Herr W, Moosmann A, Strobel J, Hackstein H, Dornmair K, Beier F, Hansmann L. Virus-reactive T cells expanded in aplastic anemia eliminate hematopoietic progenitor cells by molecular mimicry. Blood. 2024 Apr 4:143(14):1365-1378. doi: 10.1182/blood.2023023142. Epub [PubMed PMID: 38277625]
ElNahass Y, Mekky N, Abdelfattah NM, Abdelfattah R, Samra M, Fahmy OA, Fathy G, Elmetnawy W, Sabet S, Bassiouny H, Nader H, ElHaddad A, Mahmoud HK. HLA alleles, haplotypes frequencies, and their association with hematological disorders: a report from 1550 families whose patients underwent allogeneic bone marrow transplantation in Egypt. Immunogenetics. 2024 Aug:76(4):243-260. doi: 10.1007/s00251-024-01343-x. Epub 2024 Jun 21 [PubMed PMID: 38904751]
Alshaibani A, Dufour C, Risitano A, de Latour R, Aljurf M. Hepatitis-Associated Aplastic Anemia. Hematology/oncology and stem cell therapy. 2022 Jun 1:15(2):8-12. doi: 10.1016/j.hemonc.2020.10.001. Epub 2022 Jun 1 [PubMed PMID: 33197413]
Sun W, Wu Z, Lin Z, Hollinger M, Chen J, Feng X, Young NS. Macrophage TNF-α licenses donor T cells in murine bone marrow failure and can be implicated in human aplastic anemia. Blood. 2018 Dec 27:132(26):2730-2743. doi: 10.1182/blood-2018-05-844928. Epub 2018 Oct 25 [PubMed PMID: 30361263]
Sun Y, Wu C, Liu C, Wang H, Shao Z. Increased Number of Circulating Myeloid Dendritic Cell 1 in Patients with Severe Aplastic Anemia. Clinical laboratory. 2025 Apr 1:71(4):. doi: 10.7754/Clin.Lab.2025.250146. Epub [PubMed PMID: 40209793]
Virijevic M, Marjanovic I, Andjelkovic M, Jakovic L, Micic D, Bogdanovic A, Pavlovic S. Novel telomerase reverse transcriptase gene mutation in a family with aplastic anaemia. Familial cancer. 2024 Nov:23(4):635-639. doi: 10.1007/s10689-024-00399-8. Epub 2024 May 25 [PubMed PMID: 38795222]
Gurjar H, Jain A, Wangkheimayum S, Varma S, Vijayvergiya R, Malhotra P. Danazol causes significant changes in the cardiometabolic profile of patients with acquired aplastic anaemia. Blood cells, molecules & diseases. 2025 May:112():102921. doi: 10.1016/j.bcmd.2025.102921. Epub 2025 Mar 26 [PubMed PMID: 40174329]
Zhou J, Wang Q, Yu Y, Chen L, Shi J, Huang C, Zhuang R. A rare case report of severe aplastic anaemia caused by long-term use of zidovudine. BMC infectious diseases. 2024 Dec 18:24(1):1421. doi: 10.1186/s12879-024-09875-z. Epub 2024 Dec 18 [PubMed PMID: 39695951]
Level 3 (low-level) evidenceChen T, Chen Y, Bao W, Lu W. T-lymphocyte subsets and Th1/Th2 cytokines in convalescent patients with Epstein-Barr virus-associated aplastic anemia. Hematology (Amsterdam, Netherlands). 2020 Dec:25(1):11-16. doi: 10.1080/16078454.2019.1702304. Epub [PubMed PMID: 31842718]
Mendes-de-Almeida DP, Bokel JPB, Alves ADR, Vizzoni AG, Tavares ICF, Silva MST, Netto JDSB, Grinsztejn BGJ, Amado Leon LA. Clinical Presentation of Parvovirus B19 Infection in Adults Living with HIV/AIDS: A Case Series. Viruses. 2023 May 8:15(5):. doi: 10.3390/v15051124. Epub 2023 May 8 [PubMed PMID: 37243210]
Level 2 (mid-level) evidenceNasnas P, Ling J, Gerstein Y, Wang SA, Loghavi S, Hammond D, Montalban-Bravo G, Senapati J, Pemmaraju N, Corredor J, Pierce S, Roth M, Ravandi F, Cuglievan B, Kadia T, DiNardo CD. Detection of PNH Clones can Aid in the Distinction of Aplastic Anemia vs Inherited BM Failure Syndromes: A Single Center Experience and Review of the Literature. Clinical lymphoma, myeloma & leukemia. 2024 Oct:24(10):732-735. doi: 10.1016/j.clml.2024.06.001. Epub 2024 Jun 12 [PubMed PMID: 38945779]
Fattizzo B, Ireland R, Dunlop A, Yallop D, Kassam S, Large J, Gandhi S, Muus P, Manogaran C, Sanchez K, Consonni D, Barcellini W, Mufti GJ, Marsh JCW, Kulasekararaj AG. Clinical and prognostic significance of small paroxysmal nocturnal hemoglobinuria clones in myelodysplastic syndrome and aplastic anemia. Leukemia. 2021 Nov:35(11):3223-3231. doi: 10.1038/s41375-021-01190-9. Epub 2021 Mar 4 [PubMed PMID: 33664463]
Dezern AE, Borowitz MJ. ICCS/ESCCA consensus guidelines to detect GPI-deficient cells in paroxysmal nocturnal hemoglobinuria (PNH) and related disorders part 1 - clinical utility. Cytometry. Part B, Clinical cytometry. 2018 Jan:94(1):16-22. doi: 10.1002/cyto.b.21608. Epub [PubMed PMID: 29236352]
Level 3 (low-level) evidenceGoulart H, Masarova L, Mesa R, Harrison C, Kiladjian JJ, Pemmaraju N. Myeloproliferative neoplasms in the adolescent and young adult population: A comprehensive review of the literature. British journal of haematology. 2024 Jul:205(1):48-60. doi: 10.1111/bjh.19557. Epub 2024 Jun 10 [PubMed PMID: 38853641]
Chattopadhyay S, Lionel S, Selvarajan S, Devasia AJ, Korula A, Kulkarni U, Aboobacker FN, Lakshmi KM, Srivastava A, Mathews V, Abraham A, George B. Relapse and transformation to myelodysplastic syndrome and acute myeloid leukemia following immunosuppressive therapy for aplastic anemia is more common as compared to allogeneic stem cell transplantation with a negative impact on survival. Annals of hematology. 2024 Mar:103(3):749-758. doi: 10.1007/s00277-024-05621-2. Epub 2024 Jan 20 [PubMed PMID: 38242970]
Geppner AC. Aplastic anemia: A person-centered approach to diagnosis and treatment. JAAPA : official journal of the American Academy of Physician Assistants. 2025 Apr 1:38(4):18-27. doi: 10.1097/01.JAA.0000000000000195. Epub 2025 Mar 25 [PubMed PMID: 40052724]
DeZern AE, Churpek JE. Approach to the diagnosis of aplastic anemia. Blood advances. 2021 Jun 22:5(12):2660-2671. doi: 10.1182/bloodadvances.2021004345. Epub [PubMed PMID: 34156438]
Level 3 (low-level) evidenceKulasekararaj A, Cavenagh J, Dokal I, Foukaneli T, Gandhi S, Garg M, Griffin M, Hillmen P, Ireland R, Killick S, Mansour S, Mufti G, Potter V, Snowden J, Stanworth S, Zuha R, Marsh J, BSH Committee. Guidelines for the diagnosis and management of adult aplastic anaemia: A British Society for Haematology Guideline. British journal of haematology. 2024 Mar:204(3):784-804. doi: 10.1111/bjh.19236. Epub 2024 Jan 21 [PubMed PMID: 38247114]
Solimando AG, Palumbo C, Pragnell MV, Bittrich M, Argentiero A, Krebs M. Aplastic Anemia as a Roadmap for Bone Marrow Failure: An Overview and a Clinical Workflow. International journal of molecular sciences. 2022 Oct 4:23(19):. doi: 10.3390/ijms231911765. Epub 2022 Oct 4 [PubMed PMID: 36233062]
Level 3 (low-level) evidenceIbrahim M, Khodeary A, Aziz SP, Mahmoud MG, Abdel-Baset AA, Mohamed T, Sayed SA. A flow cytometric approach to compare stem cell apoptosis in aplastic anemia and hypoplastic myelodysplastic syndrome. Hematology/oncology and stem cell therapy. 2024 Jul-Sep 01:17(3):184-189. doi: 10.4103/hemoncstem.HEMONCSTEM-D-24-00008. Epub 2024 Oct 4 [PubMed PMID: 39412754]
Kennedy AL, Shimamura A. Genetic predisposition to MDS: clinical features and clonal evolution. Blood. 2019 Mar 7:133(10):1071-1085. doi: 10.1182/blood-2018-10-844662. Epub 2019 Jan 22 [PubMed PMID: 30670445]
McReynolds LJ, Rafati M, Wang Y, Ballew BJ, Kim J, Williams VV, Zhou W, Hendricks RM, Dagnall C, Freedman ND, Carter B, Strollo S, Hicks B, Zhu B, Jones K, Paczesny S, Marsh SGE, Spellman SR, He M, Wang T, Lee SJ, Savage SA, Gadalla SM. Genetic testing in severe aplastic anemia is required for optimal hematopoietic cell transplant outcomes. Blood. 2022 Aug 25:140(8):909-921. doi: 10.1182/blood.2022016508. Epub [PubMed PMID: 35776903]
Patel BA, Ghannam J, Groarke EM, Goswami M, Dillon L, Gutierrez-Rodrigues F, Rios O, Raffo DQ, Lotter J, Young NS, Hourigan CS. Detectable mutations precede late myeloid neoplasia in aplastic anemia. Haematologica. 2021 Feb 1:106(2):647-650. doi: 10.3324/haematol.2020.263046. Epub 2021 Feb 1 [PubMed PMID: 33054127]
Steensma DP. How I use molecular genetic tests to evaluate patients who have or may have myelodysplastic syndromes. Blood. 2018 Oct 18:132(16):1657-1663. doi: 10.1182/blood-2018-06-860882. Epub 2018 Sep 5 [PubMed PMID: 30185432]
Zhang L, Han X, Zhu Q, Qin Y, Jia Y. Clinical utility of hematological parameters in aplastic anemia. Scientific reports. 2025 Jan 23:15(1):2946. doi: 10.1038/s41598-025-86917-9. Epub 2025 Jan 23 [PubMed PMID: 39849087]
Level 2 (mid-level) evidenceBortolotti M, Trikha R, Salter S, Large J, Vlachodimitropoulou E, Gandhi S, Barcellini W, Johns J, Fattizzo B, Kulasekararaj A. Pregnancy in acquired bone marrow failure syndromes: Antenatal management and maternal and fetal outcomes. American journal of hematology. 2024 Aug:99(8):1647-1650. doi: 10.1002/ajh.27372. Epub 2024 May 15 [PubMed PMID: 38747646]
Georges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood advances. 2018 Aug 14:2(15):2020-2028. doi: 10.1182/bloodadvances.2018021162. Epub [PubMed PMID: 30108110]
Level 3 (low-level) evidenceYoshida N, Kojima S. Updated Guidelines for the Treatment of Acquired Aplastic Anemia in Children. Current oncology reports. 2018 Jun 30:20(9):67. doi: 10.1007/s11912-018-0716-8. Epub 2018 Jun 30 [PubMed PMID: 29961134]
Samarasinghe S, Veys P, Vora A, Wynn R. Paediatric amendment to adult BSH Guidelines for aplastic anaemia. British journal of haematology. 2018 Jan:180(2):201-205. doi: 10.1111/bjh.15066. Epub 2017 Dec 28 [PubMed PMID: 29285764]
Gutierrez-Rodrigues F, Patel BA, Groarke EM. When to consider inherited marrow failure syndromes in adults. Hematology. American Society of Hematology. Education Program. 2023 Dec 8:2023(1):548-555. doi: 10.1182/hematology.2023000488. Epub [PubMed PMID: 38066926]
Gupta V, Eapen M, Brazauskas R, Carreras J, Aljurf M, Gale RP, Hale GA, Ilhan O, Passweg JR, Ringdén O, Sabloff M, Schrezenmeier H, Socié G, Marsh JC. Impact of age on outcomes after bone marrow transplantation for acquired aplastic anemia using HLA-matched sibling donors. Haematologica. 2010 Dec:95(12):2119-25. doi: 10.3324/haematol.2010.026682. Epub 2010 Sep 17 [PubMed PMID: 20851870]
Giammarco S, Peffault de Latour R, Sica S, Dufour C, Socie G, Passweg J, Kröger N, Petersen E, Van Lint MT, Oneto R, Signori A, Bacigalupo A, European Group for Blood and Marrow Transplantation Severe Aplastic Anemia Working Party. Transplant outcome for patients with acquired aplastic anemia over the age of 40: has the outcome improved? Blood. 2018 Apr 26:131(17):1989-1992. doi: 10.1182/blood-2017-09-807859. Epub 2018 Mar 16 [PubMed PMID: 29549172]
Iftikhar R, DeFilipp Z, DeZern AE, Pulsipher MA, Bejanyan N, Burroughs LM, Kharfan-Dabaja MA, Arai S, Kassim A, Nakamura R, Saldaña BJD, Aljurf M, Hamadani M, Carpenter PA, Antin JH. Allogeneic Hematopoietic Cell Transplantation for the Treatment of Severe Aplastic Anemia: Evidence-Based Guidelines From the American Society for Transplantation and Cellular Therapy. Transplantation and cellular therapy. 2024 Dec:30(12):1155-1170. doi: 10.1016/j.jtct.2024.09.017. Epub 2024 Sep 20 [PubMed PMID: 39307421]
Level 1 (high-level) evidenceLiu L, Han B, Zhang Y, Lei M, Liu R, Lin Z, Jiao W, Zhang F, Fu R, Zhao X, Miao M, Zhang L, Wu D. First-line treatment of severe aplastic anemia: immunosuppressive therapy plus eltrombopag versus haploidentical hematopoietic stem cell transplantation, a multicenter prospective study. Bone marrow transplantation. 2024 Oct:59(10):1449-1457. doi: 10.1038/s41409-024-02377-1. Epub 2024 Aug 1 [PubMed PMID: 39090437]
Wu L, Liu L, Zhao X, Zhou M, Fu A, Zhang Y, Yang W, Chen X, Mo W, Wang C, Li Y, Xu S, Pan S, Zhou R, Meng F, Zhang F, Wu D, Wang S. Unrelated donor hematopoietic stem cell transplantation compared to immunosuppressive therapy plus eltrombopag as first-line treatment for adults with severe aplastic anemia. Blood cancer journal. 2024 Mar 6:14(1):37. doi: 10.1038/s41408-024-01021-x. Epub 2024 Mar 6 [PubMed PMID: 38443356]
Wu LQ, Huang LF, Yang H, Ye BD, Sheng JP, Yu QH, Yang Y, Jia JS, Zhang DH, Lin SY, He GS, Li JY. Comparison of haploidentical-allogeneic hematopoietic stem cell transplantation and intensive immunosuppressive therapy for patients with severe aplastic anemia with an absolute neutrophil count of zero: a retrospective study. Annals of hematology. 2023 Aug:102(8):2015-2023. doi: 10.1007/s00277-023-05256-9. Epub 2023 May 17 [PubMed PMID: 37193759]
Level 2 (mid-level) evidencePeffault de Latour R, Kulasekararaj A, Iacobelli S, Terwel SR, Cook R, Griffin M, Halkes CJM, Recher C, Barraco F, Forcade E, Vallejo JC, Drexler B, Mear JB, Smith AE, Angelucci E, Raymakers RAP, de Groot MR, Daguindau E, Nur E, Barcellini W, Russell NH, Terriou L, Iori AP, La Rocca U, Sureda A, Sánchez-Ortega I, Xicoy B, Jarque I, Cavenagh J, Sicre de Fontbrune F, Marotta S, Munir T, Tjon JML, Tavitian S, Praire A, Clement L, Rabian F, Marano L, Hill A, Palmisani E, Muus P, Cacace F, Frieri C, van Lint MT, Passweg JR, Marsh JCW, Socié G, Mufti GJ, Dufour C, Risitano AM, Severe Aplastic Anemia Working Party of the European Society for Blood and Marrow Transplantation. Eltrombopag Added to Immunosuppression in Severe Aplastic Anemia. The New England journal of medicine. 2022 Jan 6:386(1):11-23. doi: 10.1056/NEJMoa2109965. Epub [PubMed PMID: 34986284]
Patel BA, Groarke EM, Lotter J, Shalhoub R, Gutierrez-Rodrigues F, Rios O, Quinones Raffo D, Wu CO, Young NS. Long-term outcomes in patients with severe aplastic anemia treated with immunosuppression and eltrombopag: a phase 2 study. Blood. 2022 Jan 6:139(1):34-43. doi: 10.1182/blood.2021012130. Epub [PubMed PMID: 34525188]
Townsley DM, Scheinberg P, Winkler T, Desmond R, Dumitriu B, Rios O, Weinstein B, Valdez J, Lotter J, Feng X, Desierto M, Leuva H, Bevans M, Wu C, Larochelle A, Calvo KR, Dunbar CE, Young NS. Eltrombopag Added to Standard Immunosuppression for Aplastic Anemia. The New England journal of medicine. 2017 Apr 20:376(16):1540-1550. doi: 10.1056/NEJMoa1613878. Epub [PubMed PMID: 28423296]
Patel BA, Townsley DM, Scheinberg P. Immunosuppressive therapy in severe aplastic anemia. Seminars in hematology. 2022 Jan:59(1):21-29. doi: 10.1053/j.seminhematol.2022.01.002. Epub 2022 Jan 19 [PubMed PMID: 35491055]
Foucar CE, Foley DH, Aldous J, Burke PW, Pettit KR, Benitez LL, Perissinotti AJ, Marini BL, Boonstra P, Bixby DL. Real-world outcomes with immunosuppressive therapy for aplastic anemia in patients treated at the University of Michigan. European journal of haematology. 2024 Mar:112(3):424-432. doi: 10.1111/ejh.14131. Epub 2023 Nov 6 [PubMed PMID: 37929654]
Carpenedo M, Zappaterra A, Del Castello L, Ferrari B, Cotilli G, Bernasconi DP, Pezzatti S, Sacco F, Borin L, Carrer A, Verga L, Brioschi F. Feasibility and effectiveness of the prolonged use of eltrombopag in addition to immunosuppression in patients with acquired aplastic anemia: a single-center real-life experience. Platelets. 2024 Dec:35(1):2415483. doi: 10.1080/09537104.2024.2415483. Epub 2024 Oct 16 [PubMed PMID: 39641636]
Level 2 (mid-level) evidenceLi X, Shangguan X, Wang H, Wang Q, Zhang Y, Han B, Liu R, Zhang F, Fu R, Lin Z, Miao M, Ma X, Lei M, Wu D, Liu L. Comparison of efficacy of eltrombopag combined with immunosuppression in the treatment of severe aplastic anemia and very severe aplastic anemia: real-world data and evidence. Annals of hematology. 2024 Sep:103(9):3483-3491. doi: 10.1007/s00277-024-05910-w. Epub 2024 Aug 1 [PubMed PMID: 39088061]
Li J, Liang W, Fan H, Zhou K, Li Y, Yang W, Jing L, Zhang L, Ye L, Xiong Y, Peng G, Yang Y, Yuan W, Shi J, Zhang F, Zhao X. Efficacy and safety of avatrombopag in combination with standard immunosuppressive therapy for severe aplastic anemia. Experimental hematology. 2024 Dec:140():104670. doi: 10.1016/j.exphem.2024.104670. Epub 2024 Nov 5 [PubMed PMID: 39505082]
Prabahran A, Durrani J, Coelho-Da Silva J, Shalhoub R, Lotter J, Rios O, Ritchie DS, Wu CO, Patel BA, Young NS, Groarke EM. Safety and efficacy of immunosuppressive therapy for elderly patients with severe aplastic anaemia. British journal of haematology. 2024 Sep:205(3):1170-1179. doi: 10.1111/bjh.19648. Epub 2024 Jul 17 [PubMed PMID: 39021060]
Nakamura Y, Zaimoku Y, Yamaguchi H, Yamazaki H, Kanaya M, Uchida N, Doki N, Sakurai M, Hiramoto N, Kako S, Onizuka M, Onodera K, Maruyama Y, Ohigashi H, Nishida T, Yoshihara S, Matsuoka KI, Eto T, Kanda Y, Fukuda T, Atsuta Y, Onishi Y. Significance of absolute neutrophil count before allogeneic hematopoietic stem cell transplantation in adult patients with aplastic anemia. Annals of hematology. 2024 Aug:103(8):3121-3133. doi: 10.1007/s00277-024-05800-1. Epub 2024 May 16 [PubMed PMID: 38750374]
Shinn LT, Benitez LL, Perissinotti AJ, Reid JH, Buhlinger KM, van Deventer H, Barth D, Wagner CB, Zacholski K, Desai R, Soule A, Stump SE, Weis TM, Bixby D, Burke P, Pettit K, Marini BL. Multicenter evaluation of the addition of eltrombopag to immunosuppressive therapy for adults with severe aplastic anemia. International journal of hematology. 2023 Dec:118(6):682-689. doi: 10.1007/s12185-023-03670-3. Epub 2023 Oct 26 [PubMed PMID: 37882977]
Scheinberg P. Activity of eltrombopag in severe aplastic anemia. Blood advances. 2018 Nov 13:2(21):3054-3062. doi: 10.1182/bloodadvances.2018020248. Epub [PubMed PMID: 30425070]
Level 3 (low-level) evidenceScheinberg P, Nunez O, Weinstein B, Scheinberg P, Biancotto A, Wu CO, Young NS. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. The New England journal of medicine. 2011 Aug 4:365(5):430-8. doi: 10.1056/NEJMoa1103975. Epub [PubMed PMID: 21812672]
Killick SB, Bown N, Cavenagh J, Dokal I, Foukaneli T, Hill A, Hillmen P, Ireland R, Kulasekararaj A, Mufti G, Snowden JA, Samarasinghe S, Wood A, Marsh JC, British Society for Standards in Haematology. Guidelines for the diagnosis and management of adult aplastic anaemia. British journal of haematology. 2016 Jan:172(2):187-207. doi: 10.1111/bjh.13853. Epub 2015 Nov 16 [PubMed PMID: 26568159]
Scheinberg P, Young NS. How I treat acquired aplastic anemia. Blood. 2012 Aug 9:120(6):1185-96. doi: 10.1182/blood-2011-12-274019. Epub 2012 Apr 19 [PubMed PMID: 22517900]
Höchsmann B, Moicean A, Risitano A, Ljungman P, Schrezenmeier H. Supportive care in severe and very severe aplastic anemia. Bone marrow transplantation. 2013 Feb:48(2):168-73. doi: 10.1038/bmt.2012.220. Epub 2012 Dec 3 [PubMed PMID: 23208312]
Winkler T, Fan X, Cooper J, Desmond R, Young DJ, Townsley DM, Scheinberg P, Grasmeder S, Larochelle A, Desierto M, Valdez J, Lotter J, Wu C, Shalhoub RN, Calvo KR, Young NS, Dunbar CE. Treatment optimization and genomic outcomes in refractory severe aplastic anemia treated with eltrombopag. Blood. 2019 Jun 13:133(24):2575-2585. doi: 10.1182/blood.2019000478. Epub 2019 Apr 16 [PubMed PMID: 30992268]
Gutierrez-Rodrigues F, Munger E, Ma X, Groarke EM, Tang Y, Patel BA, Catto LFB, Clé DV, Niewisch MR, Alves-Paiva RM, Donaires FS, Pinto AL, Borges G, Santana BA, McReynolds LJ, Giri N, Altintas B, Fan X, Shalhoub R, Siwy CM, Diamond C, Raffo DQ, Craft K, Kajigaya S, Summers RM, Liu P, Cunningham L, Hickstein DD, Dunbar CE, Pasquini R, De Oliveira MM, Velloso EDRP, Alter BP, Savage SA, Bonfim C, Wu CO, Calado RT, Young NS. Differential diagnosis of bone marrow failure syndromes guided by machine learning. Blood. 2023 Apr 27:141(17):2100-2113. doi: 10.1182/blood.2022017518. Epub [PubMed PMID: 36542832]
Cheon H, Dziewulska KH, Moosic KB, Olson KC, Gru AA, Feith DJ, Loughran TP Jr. Advances in the Diagnosis and Treatment of Large Granular Lymphocytic Leukemia. Current hematologic malignancy reports. 2020 Apr:15(2):103-112. doi: 10.1007/s11899-020-00565-6. Epub [PubMed PMID: 32062772]
Level 3 (low-level) evidenceDietz AC, Savage SA, Vlachos A, Mehta PA, Bresters D, Tolar J, Bonfim C, Dalle JH, de la Fuente J, Skinner R, Boulad F, Duncan CN, Baker KS, Pulsipher MA, Lipton JM, Wagner JE, Alter BP. Late Effects Screening Guidelines after Hematopoietic Cell Transplantation for Inherited Bone Marrow Failure Syndromes: Consensus Statement From the Second Pediatric Blood and Marrow Transplant Consortium International Conference on Late Effects After Pediatric HCT. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2017 Sep:23(9):1422-1428. doi: 10.1016/j.bbmt.2017.05.022. Epub 2017 May 19 [PubMed PMID: 28533057]
Level 3 (low-level) evidenceRice C, Eikema DJ, Marsh JCW, Knol C, Hebert K, Putter H, Peterson E, Deeg HJ, Halkes S, Pidala J, Anderlini P, Tischer J, Kroger N, McDonald A, Antin JH, Schaap NP, Hallek M, Einsele H, Mathews V, Kapoor N, Boelens JJ, Mufti GJ, Potter V, Pefault de la Tour R, Eapen M, Dufour C. Allogeneic Hematopoietic Cell Transplantation in Patients Aged 50Years or Older with Severe Aplastic Anemia. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation. 2019 Mar:25(3):488-495. doi: 10.1016/j.bbmt.2018.08.029. Epub 2018 Sep 5 [PubMed PMID: 30194027]
Scheinberg P. Recent Advances and Long-Term Results of Medical Treatment of Acquired Aplastic Anemia: Are Patients Cured? Hematology/oncology clinics of North America. 2018 Aug:32(4):609-618. doi: 10.1016/j.hoc.2018.03.003. Epub 2018 May 18 [PubMed PMID: 30047414]
Level 3 (low-level) evidenceKuwatsuka Y, Ito H, Tabuchi K, Konuma T, Uchida N, Inamoto Y, Inai K, Nishida T, Ikegame K, Eto T, Katayama Y, Kataoka K, Tanaka M, Takahashi S, Fukuda T, Ichinohe T, Kimura F, Kanda J, Atsuta Y, Matsuo K. Trends in allogeneic hematopoietic cell transplantation survival using population-based descriptive epidemiology method: analysis of national transplant registry data. Bone marrow transplantation. 2024 Sep:59(9):1295-1301. doi: 10.1038/s41409-024-02326-y. Epub 2024 Jun 19 [PubMed PMID: 38898226]
Wei X, Zhu W, Li J, Zhou S, Zhu Q, Ma X, Han Y, Wang Y, Miao M, Qiu H, Wu D, Wu X. The Role of Pre-existing Anti-HLA Antibodies in Severe Aplastic Anemia Patients Undergoing Allogenic Hematopoietic Stem Cell Transplantation. Transplantation and cellular therapy. 2024 Sep:30(9):902.e1-902.e11. doi: 10.1016/j.jtct.2024.05.008. Epub 2024 May 11 [PubMed PMID: 38740139]
Level 2 (mid-level) evidenceZielińska P, Noster I, Wieczorkiewicz-Kabut A, Białas K, Koclęga A, Helbig G. Allogeneic hematopoietic stem cell transplantation for acquired severe aplastic anemia: a summary of a 20-year experience. Polish archives of internal medicine. 2023 Aug 30:133(7-8):. pii: 16448. doi: 10.20452/pamw.16448. Epub 2023 Feb 28 [PubMed PMID: 36861425]
José C JP, Mariana GT, Renata V BL, Olga G CR, César H GA, David GA. Outcomes of Delayed HLA Haploidentical Transplantation with Peripheral Blood Allografts for High-Risk Patients with Severe Aplastic Anemia. Revista de investigacion clinica; organo del Hospital de Enfermedades de la Nutricion. 2025:77(1):26-33. doi: 10.24875/RIC.25000012. Epub [PubMed PMID: 40048745]
Gao M, Huang X, Gao S, Wang S, Deng J, Zhang Y, Kong P, Zhang C, Gao L, Feng Y, Zhu L, Liu J, Chen T, Yao H, Wang L, Liu H, Liu Y, Zhao L, Zhang X, Gao L. Similar outcomes between HLA-haploid and matched sibling donor hematopoietic stem cell transplantation: a multicenter, retrospective study and severe aplastic anemia transplant-specific prognostic scoring system. Annals of hematology. 2025 Jan:104(1):781-791. doi: 10.1007/s00277-024-06051-w. Epub 2024 Dec 12 [PubMed PMID: 39663257]
Level 2 (mid-level) evidence