Introduction
By definition, neurogenic pulmonary edema (NPE) is a clinical condition characterized by the acute accumulation of extravascular pulmonary fluid following a severe central nervous system (CNS) insult, most commonly involving the brainstem, and typically manifests as acute respiratory distress. W. T. Shanahan was the first to describe acute NPE in 1908. Francois Moutier notably described the sudden onset of pulmonary edema among soldiers who sustained headshot wounds during World War I.
NPE requires the exclusion of other identifiable pulmonary lesions or sources of cardiovascular impairment that may accompany nervous system compromise, eg, pulmonary aspiration or other ischemic, toxic, or traumatic lesions of the heart or lungs, even though these conditions may coexist with pulmonary edema.
If the clinical presentation is unequivocal, the diagnosis should be presumed when acute pulmonary edema is associated with CNS injury in the absence of primary pulmonary or cardiovascular damage. However, some ambiguity may still persist, particularly because the exact pathogenesis of NPE is not fully understood, as described in the literature. One of the main proposed mechanisms is sympathetic storm/catecholamine-mediated pulmonary vasoconstriction.[1][2][3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The underlying causes of NPE reported in the literature include the following common and rarer etiologies:
- Common etiologies
- Acute subarachnoid hemorrhage (SAH)
- Intracerebral haemorrhage
- Traumatic brain injury (eg, subdural/epidural hematoma, brainstem or upper cervical spinal trauma)
- Epilepsy
- Rare etiologies
Any acute CNS injury may result in NPE. However, the 3 most common causes of this syndrome are open or closed cranial trauma, subarachnoid hemorrhage (SAH) following aneurysmal rupture, and epilepsy with generalized tonic-clonic seizures.[5][6][7] Pulmonary complications occur in up to 50% of aneurysmal SAH patients, with NPE being one of the more serious manifestations.[9] NPE is associated with high mortality, particularly in patients with severe brain injury or poor neurological status. Reported mortality rates vary, with some series citing figures as high as 60% to 100%.[2][3][10]
Epidemiology
The actual incidence of NPE remains unknown. However, through pathological data analysis, experts have proposed that the condition is often observed at autopsy in patients who die suddenly after a seizure, eg, in sudden, unexpected death epilepsy, and also in victims of severe head injury. Although NPE is rare in the general population, autopsy studies suggest it may be present in up to 20% of patients with severe traumatic brain injury.[11][12][13][14]
Pathophysiology
A comprehensive understanding of the pathophysiology of NPE remains uncertain (see Image. Pathophysiology of Neurogenic Pulmonary Edema). The CNS disturbance will cause a massive sympathetic discharge (or catecholamine surge), leading to systemic vasoconstriction, which increases venous return and pulmonary capillary hydrostatic pressure. That change in pressure will result in the leakage of intravascular fluid into both the alveoli and the pulmonary interstitial space through the following 2 mechanisms:
- Increased pulmonary capillary hydrostatic pressure causing fluid movement into the alveoli as a result of Starling forces
- Increased permeability of the pulmonary capillary endothelium due to catecholamine-mediated injury
For a complete, comprehensive understanding of the pathophysiology of NPE, clinicians should consider 3 different sectors: the central, autonomic, and cardiovascular and pulmonary systems. However, the fact that this clinical condition results from the overlapping of these sectors through cause and effect is worth noting.
Central Nervous System
Structural considerations
The injured CNS will initiate a state of sympathetic overactivity. Specific centers in the CNS, when stimulated, will result in activation of the autonomic sympathetic system. Centers responsible for autonomic contributions to the pathogenesis of NPE are located in specific areas of the CNS that elicit NPE. These areas are located in the rostral ventrolateral medulla, area postrema, nuclei of the solitary tract, nuclei of A1 and A5, the medial reticulated nucleus, and the dorsal motor vagus nucleus.
Chemical processes
The role of neurotransmitters in the pathogenesis of NPE is unclear. Experimental studies suggest that NMDA receptor activation enhances sympathetic output, while GABAergic inhibition may counteract this response after CNS injury.
Physiological events
Increased intracranial pressure (ICP) is a common occurrence after CNS injury. The abrupt increase in ICP will result in the Cushing triad of hypertension, irregular breathing, and bradycardia. These physiological changes, along with sympathetic overflow, promote the development of pulmonary edema. Experimental studies in animals have demonstrated an increase in pulmonary artery pressure and an increase in extravascular pulmonary fluid volume in response to ICP.
Autonomic Nervous System
Sympathetic overflow
Excessive sympathetic discharge is considered the key driver of NPE pathogenesis, leading to a catecholamine surge and subsequent cardiovascular and pulmonary changes.[15] The sudden over-activation of the NPE trigger zone, either due to direct injury or irritation, activation of ascending neural pathways, or as a response to the raised ICP, will lead to sympathetic overflow and a catecholamine release that initiates 3 important pathophysiological responses: systemic vasoconstriction, hypertension, and an increase in venous return.[16][15]
Parasympathetic contribution
Although the vagus nerve influences heart rate and bronchomotor tone, its role in NPE remains unclear. Some speculate that vagally mediated bradycardia may contribute, but no definitive causal relationship has been established. Clinicians should also note that the correlation between vagus nerve activity and the pathogenesis of NPE raises the question of whether bradycardia is an essential or an accessory factor in the development of pulmonary edema.
Cardiovascular and Pulmonary Systems
Sympathetic overflow and a catecholamine surge result in an increase in systemic resistance, venous return, and blood pressure. The proposed theories of the development of NPE include:
- Hemodynamic changes: Increased functional demand on the cardiac muscle due to the previously mentioned outcomes of the sympathetic overflow will cause the movement of blood from the systemic highly resistant circulation to the less resistant pulmonary circulation, increasing the pulmonary capillary positive hydrostatic pressure that results in the movement of fluid from the capillaries into the lung tissue and the interstitial space.
- Neurogenic myocardial injury (Takotsubo-like Cardiomyopathy): Catecholamine surge can cause neurogenic myocardial stunning, leading to transient left ventricular dysfunction, which exacerbates pulmonary congestion and edema. The increase in systemic blood pressure and venous return will cause a volume overload on the heart. As the left ventricle fails to functionally meet the loading change, accumulation of blood in the ventricle occurs, leading to cardiac damage and diastolic dysfunction. This will lead to pulmonary vascular congestion and subsequent pulmonary edema.
- Pulmonary capillary permeability: The following 2 possible causes govern increased pulmonary capillary permeability:
- Direct (humoral): Damage to the pulmonary capillary endothelium in direct response to the catecholamine release, regardless of hemodynamic changes.[17][18][19][20]
- Indirect (physical or "blast injury"): The rapid and severe elevation in pulmonary capillary hydrostatic pressure can cause direct mechanical stress and barotrauma to the delicate capillary endothelium and alveolar epithelium.[21][22][23]
History and Physical
Clinical Features
Rapid changes characterize the early stages of NPE. NPE has been reported across all age groups, but some studies suggest it may be more frequent in children and young adults following acute intracranial injury. In cases of blunt head injuries, NPE may develop within a matter of minutes. While no pathognomonic symptom defines NPE, its clinical presentation typically mirrors that of acute pulmonary edema. NPE is most often concomitant with the initial phase of the neurological pathology. Clinical signs mimic pulmonary edema but typically lack features of left ventricular failure, helping distinguish NPE from cardiogenic causes. Classic NPE typically manifests within 2 to 12 hours after neurological injury, though onset can be more rapid in severe cases.
Symptoms resemble other forms of pulmonary edema, including dyspnea, tachypnea, and impaired gas exchange, often accompanied by systolic hypertension, which may reflect intracranial hypertension. In patients with spontaneous ventilation, early signs include dyspnea, tachypnea, cough, and rales on auscultation, as well as tachycardia, which may be accompanied by pink, foamy sputum or hemoptysis. Additional sympathetic signs, such as diaphoresis, transient hypertension, paralytic ileus, and insomnia, may also be observed. Ventilation and perfusion disorders, hypoxemia, and carbon dioxide retention frequently occur shortly after that.
If untreated, the natural evolution of NPE leads to respiratory failure, followed by cardiovascular collapse, and has a high mortality rate that has been estimated at more than 60%. In many cases, determining whether mortality is primarily due to the pulmonary, neurological, or cardiovascular components of the illness is difficult. In contrast to older descriptions, more recent publications seem to attribute mortality to the brain injury. However, in cases of pediatric encephalomyelitis, the cause of death would be predominantly pulmonary or cardiovascular failure.[2][24][25]
Evaluation
NPE is a clinical diagnosis made in the context of acute CNS injury when pulmonary symptoms are present and other causes of pulmonary edema have been excluded. The primary aim of diagnostic studies is to rule out alternative causes of pulmonary edema, eg, cardiogenic, infectious, or traumatic etiologies.
Radiographic Studies
A chest x-ray (CXR) is essential for differentiating this condition from aspiration pneumonitis. With aspiration pneumonitis, the radiographic features will evolve over a few hours and can take up to 3 weeks to resolve. This pattern contrasts with the alveolar infiltrates seen in NPE, which typically occur immediately after the injury. Excluding other pulmonary causes, including lung contusion, hemothorax, and pneumothorax, is necessary to confirm the diagnosis of NPE.
Electrocardiography and Echocardiography
Electrocardiography and echocardiography are essential to assess for myocardial ischemia, valvular abnormalities, or left ventricular dysfunction that may indicate cardiogenic edema.
Biomarkers
Brain natriuretic peptide (BNP) and troponin levels may be measured to assess for concurrent cardiogenic causes, but are not specific to NPE. Cardiac enzymes (eg, troponins) can help identify myocardial injury and assist in distinguishing NPE from acute coronary syndromes.[26][27]
Treatment / Management
With appropriate treatment, NPE typically resolves within 48 to 72 hours, after which the severity of the underlying neurological injury largely determines the overall prognosis. Severe cases of NPE have a high mortality, requiring intensive care. If NPE is clinically suspected, the initial management involves oxygen supplementation, artificial ventilation, and intensive care monitoring if needed. Consideration should be given to continually measuring ICP, and pulmonary artery catheterization may be considered in selected cases to assess pulmonary capillary wedge pressure (PCWP).
Definitive treatment should target the underlying CNS insult, though this may be challenging in cases of severe traumatic brain injury (TBI). The primary goal in managing diffuse injury to the CNS is to reduce ICP. Based on current pathophysiological hypotheses, cardiovascular therapy aims to increase the inotropic response through beta-adrenergic stimulation, and efforts to reduce pulmonary vascular resistance are essential. Nitric oxide, used for this purpose, is experimental but may have beneficial effects. In the later stages of NPE, the cardiovascular and ventilation strategies used in acute respiratory distress syndrome (ARDS) will be required.[28] The administration of steroids remains controversial.[29][30][31](A1)
Differential Diagnosis
Differential diagnoses that should also be considered when evaluating a patient with suspected NPE include:
- Adult respiratory distress syndrome
- Aspiration pneumonitis and pneumonia
- Bacterial pneumonia
- Cardiogenic pulmonary edema (see Table. Differentiating Neurogenic Pulmonary Edema and Cardiogenic Pulmonary Edema)
- SAH
Table. Differentiating Neurogenic Pulmonary Edema and Cardiogenic Pulmonary Edema
|
Feature |
NPE |
CPE |
|
Primary Trigger |
Acute, severe CNS insult (TBI, SAH, seizure, etc.) |
Primary cardiac dysfunction (left ventricular failure, valvular disease, arrhythmia) or conditions causing high cardiac filling pressures |
|
Primary Pathophysiology |
Sympathetic surge, catecholamine release, "blast injury," increased pulmonary capillary permeability & hydrostatic pressure |
Increased left ventricle (LV) filling pressures, increased pulmonary venous pressure, increased pulmonary capillary hydrostatic pressure; later, possible increased permeability ("stress failure") |
|
Onset |
Rapid (minutes to hours after CNS insult) |
Can be acute ("flash") or gradual worsening |
|
Key History |
Recent severe CNS injury/event |
History of heart disease, orthopnea, paroxysmal nocturnal dyspnea |
|
Clinical Signs |
Respiratory distress, pink frothy sputum, normal JVP (usually), signs of CNS injury, possible hypertension |
Respiratory distress, orthopnea, basal crackles, ±S3 gallop, ±elevated jugular venous pressure, ±peripheral edema, signs of cardiac cause |
|
Chest X-Ray Findings |
Bilateral, diffuse/homogeneous opacities, often apical predominance, normal heart size, rapid resolution with CNS improvement |
Perihilar "batwing" edema, cephalization, Kerley B lines, pleural effusions, cardiomegaly |
|
Echocardiography |
Usually normal LV function (or transient Takotsubo-like dysfunction), no primary valvular disease, no LA hypertension |
LV systolic/diastolic dysfunction, valvular disease, wall motion abnormalities, signs of elevated left atrial pressure |
|
BNP/NT-proBNP |
Normal or mildly elevated (unless significant neurogenic myocardial stunning) |
Markedly elevated |
|
Cardiac Enzymes |
May be elevated (catecholamine-induced myocardial injury) |
Often elevated (if acute coronary syndrome-related or severe heart failure) |
|
PAWP |
Normal (<18 mm Hg) |
Elevated (>18 mm Hg) |
|
Edema Fluid |
Often protein-rich (exudative features due to permeability) |
Initially protein-poor (transudate), can become protein-rich with "stress failure |
Prognosis
The prognosis of NPE is closely tied to the severity of the underlying central nervous system injury rather than the pulmonary condition itself. While the respiratory manifestations of NPE can be severe and may rapidly progress to respiratory failure and cardiovascular compromise in some cases, appropriate intensive care and supportive management often lead to significant improvement within 48 to 72 hours.
Despite this potential for recovery, the overall mortality rate remains high, estimated at over 60% in some studies, particularly in cases involving extensive brain injury. Outcomes are generally poorer in severe traumatic brain injuries and pediatric encephalomyelitis, where pulmonary and cardiovascular complications contribute significantly to mortality.
Complications
NPE can lead to several life-threatening complications, primarily stemming from rapid respiratory and cardiovascular deterioration. Acute respiratory failure is a common consequence, often requiring mechanical ventilation and intensive care. Cardiovascular complications, including systemic hypertension, neurogenic myocardial injury, and eventual cardiovascular collapse, further worsen the clinical picture.
Hypoxemia and ventilation-perfusion mismatch can develop quickly, exacerbating neurological injury through secondary brain hypoxia. Additionally, the high pulmonary capillary pressures and increased permeability may cause long-term lung injury if not promptly managed. The overlapping impact of pulmonary, cardiac, and neurological dysfunction makes NPE a condition with a high risk of multiorgan failure and death.
Deterrence and Patient Education
Prevention of NPE primarily focuses on the prompt recognition and management of CNS injuries to prevent secondary complications. Early control of intracranial pressure, vigilant cardiovascular monitoring, and immediate supportive respiratory care are essential strategies. Although patient education is limited due to the acute and often sudden nature of NPE, informing families and caregivers about the signs of respiratory distress in patients with recent CNS trauma is essential. Additionally, interprofessional communication among clinicians is critical to ensure timely diagnosis and intervention, ultimately improving outcomes and reducing the risk of severe complications associated with NPE.
Pearls and Other Issues
Key factors that should be kept in mind in the management of NPE include:
- Fulminant cases of NPE have been reported in children with severe Hand, Foot, and Mouth Disease caused by enterovirus 71, likely due to brainstem involvement.[32]
- Aspiration pneumonia and NPE are distinct conditions that require different treatments. The focal point of the treatment involves finding a balance between measures such as reducing intracranial pressure, optimizing oxygenation, decreasing preload and afterload, and improving cardiac contractility.
- NPE often occurs for a short time (minutes to 48 hours) after the CNS injury and is mainly manifested by acute respiratory failure. The clinical diagnosis is easy to determine in young patients without a history of cardio-respiratory disorders or direct lesions of these organs. NPE can be complicated in polytrauma patients or older people with preexisting cardiac or pulmonary insufficiency.
- Phentolamine, phenoxybenzamine, and other alpha-receptor blocking agents have been trialed in experimental and limited clinical settings, with some reports suggesting potential benefit in mitigating catecholamine-mediated effects.[33][34]
- Droperidol is theoretically proposed as a useful agent due to its alpha-receptor-blocking properties and the ability to reduce cerebral metabolism, though clinical evidence is limited.
- In cases where severe depression of myocardial function is present, dobutamine has been used to reverse this dysfunction.
- While fluid restriction and diuretics are good options, clinicians should be cautious with their applications because some patients may be hypovolemic.
Enhancing Healthcare Team Outcomes
The diagnosis and management of NPE require a high level of clinical skill and interprofessional collaboration, given the condition’s ability to mimic other pulmonary pathologies and the absence of a specific diagnostic marker. Physicians, including internists, cardiologists, intensivists, pulmonologists, and nephrologists, must apply strong clinical judgment to differentiate NPE from conditions such as aspiration pneumonia, pulmonary embolism, or cardiogenic pulmonary edema. Advanced practitioners play an essential role in the early identification of respiratory distress and in initiating appropriate diagnostic workups. Nurses are critical for continuous bedside monitoring, recognizing subtle changes in neurological or respiratory status, and ensuring timely interventions. Pharmacists contribute by guiding safe medication use, particularly in the judicious administration of diuretics, considering that many patients may be hypovolemic and require careful fluid management rather than aggressive diuresis.
Effective interprofessional communication and coordinated care strategies are key to enhancing patient-centered care, improving outcomes, ensuring patient safety, and optimizing team performance in managing NPE. Real-time sharing of clinical findings and collaborative treatment planning are crucial for avoiding delays in diagnosis and ensuring comprehensive care, which encompasses not only respiratory and cardiovascular support but also the prevention of secondary complications, such as deep vein thrombosis and pressure ulcers. Each professional must understand their specific responsibilities and maintain open channels of communication to adjust treatment as the patient’s condition evolves. Coordinated, interprofessional efforts enable a holistic management plan that addresses the primary neurological injury, optimizes cardiopulmonary function, and reduces the risk of morbidity and mortality, ultimately achieving better recovery trajectories for patients with NPE.
Media
(Click Image to Enlarge)
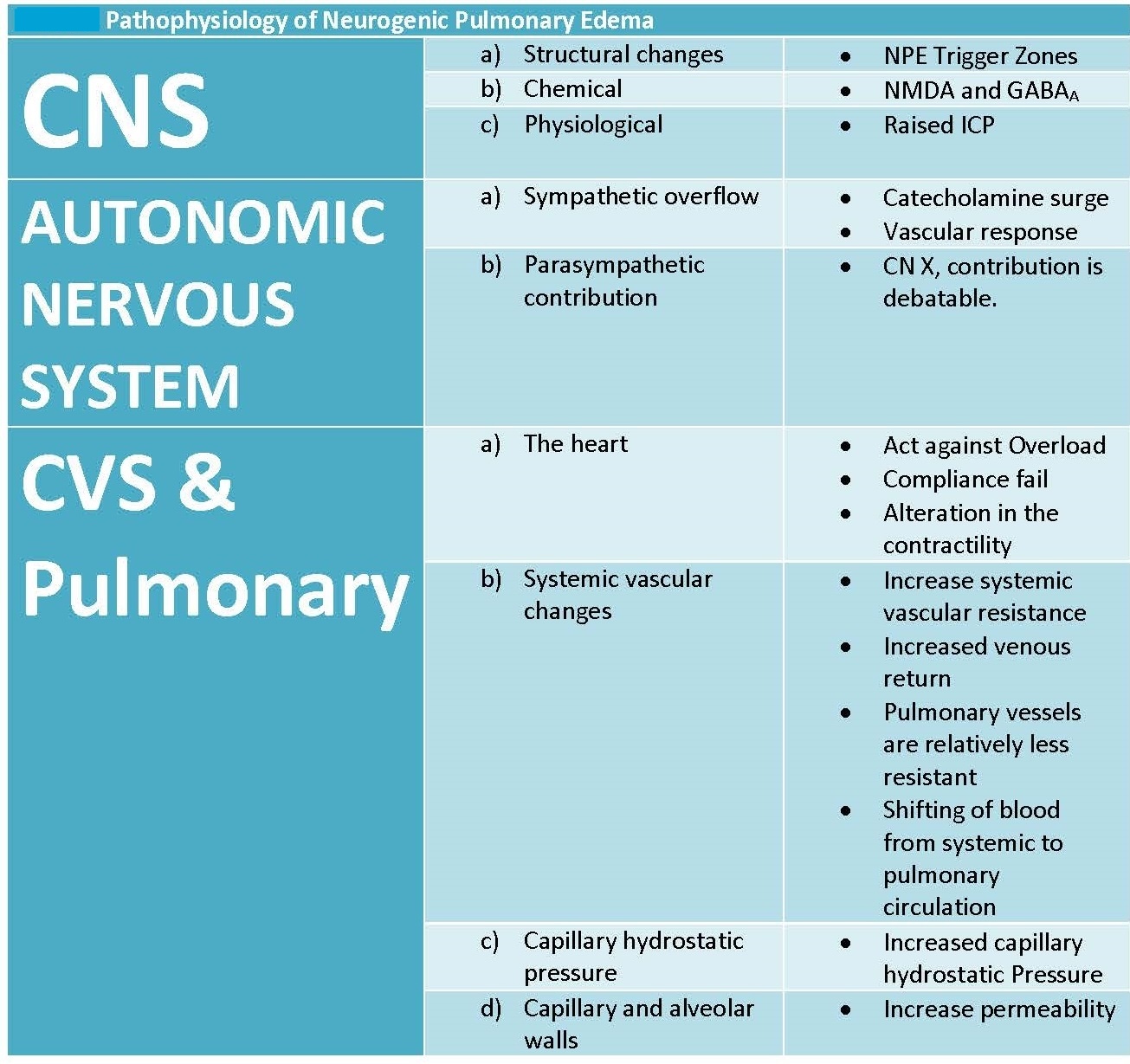
Pathophysiology of Neurogenic Pulmonary Edema. This table details the key features seen in the pathophysiology of neurogenic pulmonary edema.
Contributed by M Al-Dhahir, MD
References
Kennedy JD, Hardin KA, Parikh P, Li CS, Seyal M. Pulmonary edema following generalized tonic clonic seizures is directly associated with seizure duration. Seizure. 2015 Apr:27():19-24 [PubMed PMID: 25844030]
Raja HM, Herwadkar AV, Paroutoglou K, Lilleker JB. Neurogenic pulmonary oedema complicating a lateral medullary infarct. BMJ case reports. 2018 Jul 26:2018():. pii: bcr-2018-225437. doi: 10.1136/bcr-2018-225437. Epub 2018 Jul 26 [PubMed PMID: 30054324]
Level 3 (low-level) evidenceRomero Osorio OM, Abaunza Camacho JF, Sandoval Briceño D, Lasalvia P, Narino Gonzalez D. Postictal neurogenic pulmonary edema: Case report and brief literature review. Epilepsy & behavior case reports. 2018:9():49-50. doi: 10.1016/j.ebcr.2017.09.003. Epub 2017 Sep 28 [PubMed PMID: 29692972]
Level 3 (low-level) evidenceForgiarini EA, Cerezoli MT, Medeiros AK, Magalhães Filho MAF, Costa FMD. A new trigger for an old problem-neurogenic pulmonary edema related to intrathecal chemotherapy with pemetrexed. Jornal brasileiro de pneumologia : publicacao oficial da Sociedade Brasileira de Pneumologia e Tisilogia. 2023 May 15:49(2):e20220469. doi: 10.36416/1806-3756/e20220469. Epub 2023 May 15 [PubMed PMID: 37194815]
Miyahara M, Osaki K. Neurogenic pulmonary oedema and haemorrhage in childhood epileptic seizures: A case report and literature review. Journal of paediatrics and child health. 2023 Mar:59(3):577-579. doi: 10.1111/jpc.16382. Epub 2023 Feb 15 [PubMed PMID: 36789585]
Level 3 (low-level) evidenceGuo L, Yang X, Yang B, Tang G, Li C. Prevalence, in-hospital mortality, and factors related to neurogenic pulmonary edema after spontaneous subarachnoid hemorrhage: a systematic review and meta-analysis. Neurosurgical review. 2023 Jul 11:46(1):169. doi: 10.1007/s10143-023-02081-6. Epub 2023 Jul 11 [PubMed PMID: 37432487]
Level 1 (high-level) evidenceEnomoto T, Sekiya R, Sasaki Y, Hosoe T, Fujimoto H, Tsukamoto R, Yoshizaki A, Terashita T, Kamada H, Nakata K. Recurrent Neurogenic Pulmonary Edema Associated With Epileptic Seizures. Respirology case reports. 2025 Feb:13(2):e70108. doi: 10.1002/rcr2.70108. Epub 2025 Feb 11 [PubMed PMID: 39944276]
Level 3 (low-level) evidenceZhao J, Xuan NX, Cui W, Tian BP. Neurogenic pulmonary edema following acute stroke: The progress and perspective. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2020 Oct:130():110478. doi: 10.1016/j.biopha.2020.110478. Epub 2020 Jul 30 [PubMed PMID: 32739737]
Level 3 (low-level) evidenceFriedman JA, Pichelmann MA, Piepgras DG, McIver JI, Toussaint LG 3rd, McClelland RL, Nichols DA, Meyer FB, Atkinson JL, Wijdicks EF. Pulmonary complications of aneurysmal subarachnoid hemorrhage. Neurosurgery. 2003 May:52(5):1025-31; discussion 1031-2 [PubMed PMID: 12699543]
Felman AH. Neurogenic pulmonary edema. Observations in 6 patients. The American journal of roentgenology, radium therapy, and nuclear medicine. 1971 Jun:112(2):393-6 [PubMed PMID: 5581250]
Simmons RL, Martin AM Jr, Heisterkamp CA 3rd, Ducker TB. Respiratory insufficiency in combat casualties. II. Pulmonary edema following head injury. Annals of surgery. 1969 Jul:170(1):39-44 [PubMed PMID: 5789528]
Holland MC, Mackersie RC, Morabito D, Campbell AR, Kivett VA, Patel R, Erickson VR, Pittet JF. The development of acute lung injury is associated with worse neurologic outcome in patients with severe traumatic brain injury. The Journal of trauma. 2003 Jul:55(1):106-11 [PubMed PMID: 12855888]
Bratton SL, Davis RL. Acute lung injury in isolated traumatic brain injury. Neurosurgery. 1997 Apr:40(4):707-12; discussion 712 [PubMed PMID: 9092843]
Level 2 (mid-level) evidenceAtkinson JL. Acute lung injury in isolated traumatic brain injury. Neurosurgery. 1997 Nov:41(5):1214-6 [PubMed PMID: 9361081]
Level 3 (low-level) evidenceYang A, Liu B, Inoue T. Role of autonomic system imbalance in neurogenic pulmonary oedema. The European journal of neuroscience. 2022 Mar:55(6):1645-1657. doi: 10.1111/ejn.15648. Epub 2022 Mar 20 [PubMed PMID: 35277906]
Maslonka MA, Sheehan KN, Datar SV, Vachharajani V, Namen A. Pathophysiology and Management of Neurogenic Pulmonary Edema in Patients with Acute Severe Brain Injury. Southern medical journal. 2022 Oct:115(10):784-789. doi: 10.14423/SMJ.0000000000001457. Epub [PubMed PMID: 36191916]
Kerr NA, de Rivero Vaccari JP, Abbassi S, Kaur H, Zambrano R, Wu S, Dietrich WD, Keane RW. Traumatic Brain Injury-Induced Acute Lung Injury: Evidence for Activation and Inhibition of a Neural-Respiratory-Inflammasome Axis. Journal of neurotrauma. 2018 Sep 1:35(17):2067-2076. doi: 10.1089/neu.2017.5430. Epub 2018 Jun 8 [PubMed PMID: 29648974]
Lou M, Chen X, Wang K, Xue Y, Cui D, Xue F. Increased intracranial pressure is associated with the development of acute lung injury following severe traumatic brain injury. Clinical neurology and neurosurgery. 2013 Jul:115(7):904-8. doi: 10.1016/j.clineuro.2012.09.001. Epub 2012 Sep 23 [PubMed PMID: 23010612]
Colice GL. Neurogenic pulmonary edema. Clinics in chest medicine. 1985 Sep:6(3):473-89 [PubMed PMID: 3907948]
Level 3 (low-level) evidenceSimon RP. Neurogenic pulmonary edema. Neurologic clinics. 1993 May:11(2):309-23 [PubMed PMID: 8316188]
Level 3 (low-level) evidenceDucker TB, Simmons RL. Increased intracranial pressure and pulmonary edema. 2. The hemodynamic response of dogs and monkeys to increased intracranial pressure. Journal of neurosurgery. 1968 Feb:28(2):118-23 [PubMed PMID: 4966167]
Level 3 (low-level) evidenceBahloul M, Chaari AN, Kallel H, Khabir A, Ayadi A, Charfeddine H, Hergafi L, Chaari AD, Chelly HE, Ben Hamida C, Rekik N, Bouaziz M. Neurogenic pulmonary edema due to traumatic brain injury: evidence of cardiac dysfunction. American journal of critical care : an official publication, American Association of Critical-Care Nurses. 2006 Sep:15(5):462-70 [PubMed PMID: 16926367]
Mayer SA, Lin J, Homma S, Solomon RA, Lennihan L, Sherman D, Fink ME, Beckford A, Klebanoff LM. Myocardial injury and left ventricular performance after subarachnoid hemorrhage. Stroke. 1999 Apr:30(4):780-6 [PubMed PMID: 10187879]
Level 2 (mid-level) evidenceBean JW, Beckman DL. Centrogenic pulmonary pathology in mechanical head injury. Journal of applied physiology. 1969 Dec:27(6):807-12 [PubMed PMID: 5353203]
Level 3 (low-level) evidenceHall A, O'Kane R. The Extracranial Consequences of Subarachnoid Hemorrhage. World neurosurgery. 2018 Jan:109():381-392. doi: 10.1016/j.wneu.2017.10.016. Epub 2017 Oct 16 [PubMed PMID: 29051110]
Kitagawa T, Yamamoto J, Kureshima M, Maeda H, Nishizawa S. [Takotsubo Cardiomyopathy and Neurogenic Pulmonary Edema Following Fibrinolytic Therapy for Embolic Stroke:A Case Report]. No shinkei geka. Neurological surgery. 2018 Jan:46(1):21-25. doi: 10.11477/mf.1436203669. Epub [PubMed PMID: 29362281]
Level 3 (low-level) evidenceBonello M, Pullicino R, Larner AJ. Acute pulmonary oedema: not always cardiogenic. The journal of the Royal College of Physicians of Edinburgh. 2017 Mar:47(1):57-59. doi: 10.4997/JRCPE.2017.112. Epub [PubMed PMID: 28569284]
Fan TH, Huang M, Gedansky A, Price C, Robba C, Hernandez AV, Cho SM. Prevalence and Outcome of Acute Respiratory Distress Syndrome in Traumatic Brain Injury: A Systematic Review and Meta-Analysis. Lung. 2021 Dec:199(6):603-610. doi: 10.1007/s00408-021-00491-1. Epub 2021 Nov 15 [PubMed PMID: 34779897]
Level 1 (high-level) evidenceSARNOFF SJ, BERGLUND E, SARNOFF LC. Neurohemodynamics of pulmonary edema. III. Estimated changes in pulmonary blood volume accompanying systemic vasoconstriction and vasodilation. Journal of applied physiology. 1953 Jan:5(7):367-74 [PubMed PMID: 13022604]
Horton JM. The anaesthetist's contribution to the care of head injuries. British journal of anaesthesia. 1976 Aug:48(8):767-71 [PubMed PMID: 779815]
Herbst C, Tippler B, Shams H, Simmet T. A role for endothelin in bicuculline-induced neurogenic pulmonary oedema in rats. British journal of pharmacology. 1995 Jul:115(5):753-60 [PubMed PMID: 8548173]
Level 3 (low-level) evidencePeng L, Luo R, Jiang Z. Risk factors for neurogenic pulmonary edema in patients with severe hand, foot, and mouth disease: A meta-analysis. International journal of infectious diseases : IJID : official publication of the International Society for Infectious Diseases. 2017 Dec:65():37-43. doi: 10.1016/j.ijid.2017.09.020. Epub 2017 Sep 29 [PubMed PMID: 28970089]
Level 1 (high-level) evidenceKrishnamoorthy V, Weinberg G. Phentolamine for neurogenic pulmonary edema: bench to bedside progress. Chest. 2012 Sep:142(3):809. doi: 10.1378/chest.12-0855. Epub [PubMed PMID: 22948592]
Davison DL, Chawla LS, Selassie L, Tevar R, Junker C, Seneff MG. Neurogenic pulmonary edema: successful treatment with IV phentolamine. Chest. 2012 Mar:141(3):793-795. doi: 10.1378/chest.11-0789. Epub [PubMed PMID: 22396565]