Introduction
Pemphigus herpetiformis is a rare subtype of intraepidermal autoimmune bullous disease characterized by the clinical features of dermatitis herpetiformis and the immunologic profile of pemphigus. Although Jablonska and colleagues introduced the term pemphigus herpetiformis in 1975, earlier reports described similar presentations under various names, including dermatitis herpetiformis with acantholysis, mixed bullous disease, and pemphigus controlled by sulfapyridine.[1][2][3][4]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
Pemphigus herpetiformis appears to be mediated by an immunoglobulin G (IgG) autoantibody that primarily targets desmoglein (Dsg) proteins in the epidermis. The mechanism by which these autoantibodies produce characteristic skin lesions remains debated.[5] Some cases of drug-induced pemphigus herpetiformis have also been reported.[6][7][8][9] Recent case studies have revealed a possible association between pemphigus herpetiformis and gastric cancer.[10]
Epidemiology
Pemphigus herpetiformis accounts for 6% to 7.3% of pemphigus cases. Since the condition was initially described by Jablonska and colleagues in 1975, approximately 100 cases have been documented in the literature. Both sexes are affected equally. Reported ages of onset range from 5 to 92 years, with only 4 pediatric cases identified. No ethnic predilection has been observed. Cases have been reported across multiple continents, including North and South America, Europe, Asia, and Africa.
Pathophysiology
The primary target antigens in pemphigus herpetiformis are Dsg1 and, less frequently, Dsg3. However, recent reports have described cases lacking detectable anti-Dsg1 or -Dsg3 autoantibodies, with some patients exhibiting reactivity against alternative antigens such as desmocollins (Dsc).
Unlike pemphigus vulgaris and pemphigus foliaceus, pemphigus herpetiformis is characterized by marked inflammation, often without evident acantholysis. Autoantibodies in pemphigus herpetiformis may recognize functionally nonpathogenic epitopes on Dsg1 or Dsg3, which may explain the absence of acantholysis. A recent study supports this hypothesis, demonstrating a broader epitope distribution than that observed in pemphigus vulgaris or pemphigus foliaceus. Autoantibody-mediated stimulation of keratinocytes appears to trigger interleukin-8 release, promoting eosinophil and neutrophil recruitment. The resulting eosinophilic or neutrophilic spongiosis and focal intercellular edema contribute to blister formation.[11][12] Additionally, recent case reports have described pemphigus foliaceus transitioning to pemphigus herpetiformis.[13]
Histopathology
Histopathological features of pemphigus herpetiformis differ from those of pemphigus vulgaris and pemphigus foliaceus due to prominent spongiosis, typically without acantholysis. Histological patterns may vary among patients, and a single patient can exhibit different histologic findings over time. Multiple biopsies may therefore be required to establish the diagnosis.
The hallmark histopathologic feature is eosinophilic spongiosis. Additional findings may include neutrophilic spongiosis, mixed eosinophilic-neutrophilic spongiosis, subcorneal pustules, and intraepidermal vesicles containing eosinophils or neutrophils.[14]
Acantholysis is generally absent in early stages but may develop later in the disease course. Dermal papillary neutrophilic microabscesses may also be observed. The inflammatory cell profile varies, with mixed eosinophil and neutrophil infiltrates in 60% of cases, eosinophil-dominant infiltrates in approximately 20%, and neutrophil-dominant infiltrates in 20%.
History and Physical
Pemphigus herpetiformis typically has a subacute onset. Diagnosis is rarely established at the initial consultation due to atypical clinical features. Common differential considerations include dermatitis herpetiformis, bullous pemphigoid, and linear immunoglobulin A (IgA) bullous dermatosis.
Cutaneous findings may include erythematous, vesicular, bullous, pustular, or papular lesions arranged in a herpetiform pattern, often accompanied by intense pruritus. An annular distribution is frequently observed, likely reflecting centrifugal spread of the inflammatory process (see Image. Pemphigus Herpetiformis). In some cases, urticarial pruritic papules and plaques predominate—features uncommon in pemphigus vulgaris and pemphigus foliaceus.
Lesions typically involve the trunk and proximal extremities. Although widespread involvement is common, reports exist of localized or atypical presentations. Mucosal involvement is infrequent.
During the disease course, pemphigus herpetiformis may transition into pemphigus vulgaris or pemphigus foliaceus. Reverse transformation from other pemphigus variants to pemphigus herpetiformis has also been observed.
Evaluation
Peripheral blood eosinophilia may be present in some patients with pemphigus herpetiformis. Direct immunofluorescence of perilesional skin biopsy specimens typically reveals intercellular IgG and C3 deposition in the superficial layers of the epidermis, consistent with other pemphigus variants. Deposits may also appear in the lower epidermis in individuals with circulating autoantibodies against Dsg3.
Indirect immunofluorescence using substrates such as healthy human skin, monkey or guinea pig esophagus, or rat bladder may demonstrate intercellular IgG antibody binding. Enzyme-linked immunosorbent assay (ELISA) and immunoblotting often detect circulating antibodies targeting Dsg1 and, less frequently, Dsg3. Additional target antigens reported in recent studies include Dsc1, Dsc3, a 178-kDa antigen, the C-terminus of BP180, and the γ2 subunit of laminin 332.[15][16]
Treatment / Management
Pemphigus herpetiformis generally follows an indolent clinical course and poses less risk to life than pemphigus vulgaris or pemphigus foliaceus. Progression to these classical forms has been reported in some cases. Limited case numbers constrain the available evidence on optimal management strategies. Dapsone and corticosteroids remain the primary agents used, based on the literature review. Dapsone is widely considered the first-line treatment due to its inhibitory effects on neutrophil chemotaxis.
Corticosteroids are also effective, with lower daily doses typically sufficient to induce remission compared to other pemphigus subtypes. Dapsone and corticosteroids may be administered either as monotherapy or in combination.
Patients with refractory disease or transformation to pemphigus vulgaris or pemphigus foliaceus may require escalation to more intensive immunosuppressive regimens. Reported treatments in such cases include cyclophosphamide, mycophenolate mofetil, sulfapyridine, azathioprine, methotrexate, intravenous immunoglobulin, rituximab, and plasmapheresis.[17][18][19][20](B3)
Differential Diagnosis
Accurate diagnosis of pemphigus herpetiformis requires distinction from a wide range of primary and secondary bullous disorders. Clinical, histologic, and immunologic overlap may complicate differentiation, especially with the following disorders that share similar pruritic or vesiculobullous features:
- Bullous pemphigoid
- Erythema multiforme
- Excoriation disorder
- Familial benign pemphigus (Hailey-Hailey disease)
- IgA pemphigus
- Linear IgA bullous dermatosis
- Papular urticaria
- Paraneoplastic pemphigus
- Pemphigus erythematosus
- Pemphigus foliaceus
- Scabies
Overlapping features can obscure the diagnosis without thorough assessment. A structured approach to the differential diagnosis is essential for early and effective intervention.
Prognosis
The prognosis of pemphigus herpetiformis is generally favorable compared to classic pemphigus variants. Most studies report a high rate of therapeutic response, with 70% to 80% of patients achieving partial or complete remission, often with dapsone monotherapy or low-dose systemic corticosteroids. The clinical course is typically chronic but less aggressive, and long-term control is achievable in most cases. Mucosal involvement is rare, and extensive erosive disease is uncommon, contributing to the lower burden of morbidity.
Mortality directly attributable to pemphigus herpetiformis is exceedingly rare. Estimated death rates range from 0% to 2%, with fatalities primarily resulting from treatment complications or misdiagnosis leading to inappropriate management. In contrast to pemphigus vulgaris, where historical mortality exceeded 75% prior to the introduction of corticosteroids, pemphigus herpetiformis follows a markedly more indolent course.
Approximately 10% to 15% of cases may evolve into pemphigus foliaceus or pemphigus vulgaris, likely reflecting an immunologic shift rather than true disease transformation. Relapses may occur, particularly during tapering of therapy, but are typically milder than in other pemphigus forms. The long-term prognosis is excellent with early recognition and appropriate treatment. Many patients maintain stable remission with minimal or no systemic immunosuppression.
Complications
The most frequent complications of pemphigus herpetiformis are iatrogenic, arising from prolonged immunosuppressive therapy rather than from the disease itself. Chronic corticosteroid use may result in systemic effects such as osteoporosis, hyperglycaemia, hypertension, and increased susceptibility to infections. Secondary bacterial infections of excoriated or vesiculated skin, particularly with Staphylococcus aureus or Streptococcus pyogenes, can occur in inadequately treated or severely pruritic lesions. Although mucosal involvement is uncommon, its presence may lead to diagnostic confusion and inappropriate therapy, delaying effective disease control.
Dapsone-related adverse effects may complicate treatment. Manifestations include methemoglobinemia, hemolytic anaemia (especially in individuals with glucose-6-phosphate dehydrogenase deficiency), and dapsone hypersensitivity syndrome. Such reactions may necessitate discontinuation of an otherwise highly effective therapy.
In rare cases, pemphigus herpetiformis may immunologically transition into other pemphigus variants, such as pemphigus foliaceus. This shift may require escalation of therapy and closer clinical monitoring.
Persistent pruritus and postinflammatory dyspigmentation may affect quality of life. However, unlike pemphigus vulgaris, systemic complications such as fluid-electrolyte imbalance, protein loss, or sepsis are uncommon. With early diagnosis and careful therapeutic monitoring, the risk of serious complications remains low and primarily treatment-related.
Deterrence and Patient Education
Deterrence in pemphigus herpetiformis focuses on early symptom recognition, flare reduction, and minimization of treatment-related morbidity rather than on primary prevention, which remains unfeasible given the autoimmune and idiopathic nature of the disease. Patient education should begin at diagnosis, emphasizing the chronic, relapsing course, the need for long-term follow-up, and the importance of adherence to therapy.
Individuals should be taught to recognize early signs of relapse, including recurrent pruritic vesicles or urticarial plaques that may precede overt lesions. Patients must also understand the potential adverse effects of therapy, particularly the hematologic risks of dapsone and systemic complications associated with corticosteroids and immunosuppressants.
Instruction on wound care, infection prevention, and photoprotection may help limit secondary complications. Psychological support should be offered, given the distressing nature of visible and pruritic lesions, which can significantly impair quality of life. Where feasible, interprofessional care involving dermatologists, pharmacists, and primary care clinicians improves adherence and reduces iatrogenic harm.
Genetic counselling is not routinely indicated. However, patients should be reassured that pemphigus herpetiformis is neither contagious nor inherited in a Mendelian pattern, helping to reduce stigma and support self-efficacy in disease management.
Pearls and Other Issues
Pemphigus herpetiformis is a rare pemphigus subtype characterized by atypical clinical, histological, and immunopathological features. The underlying pathogenic mechanisms remain incompletely understood. Diagnosis is often delayed due to its unusual presentation and low prevalence. Intense pruritus, urticarial plaques, vesiculobullous eruptions in a herpetiform pattern, and absence of mucosal involvement should raise suspicion for pemphigus herpetiformis. The condition generally has a favorable prognosis and responds well to dapsone or low-dose corticosteroids, although a subset of cases may evolve into pemphigus vulgaris or pemphigus foliaceus.
Enhancing Healthcare Team Outcomes
Pemphigus herpetiformis is a rare blistering skin disorder best addressed by an interprofessional team composed of clinicians, dermatologic specialists, nurses, and pharmacists. No standardized treatment protocols have been established, though many patients respond to dapsone and corticosteroids. Other immunosuppressive and biologic agents are also used with variable success. Patient education regarding potential adverse effects is essential, as these risks often impact treatment adherence. Nurses and pharmacists play a key role in counseling, monitoring for side effects, and communicating concerns to prescribers. Pharmacists also verify dosing accuracy and perform medication reconciliation to identify potential drug interactions. This interprofessional approach improves therapeutic safety and clinical outcomes in pemphigus herpetiformis.
Media
(Click Image to Enlarge)
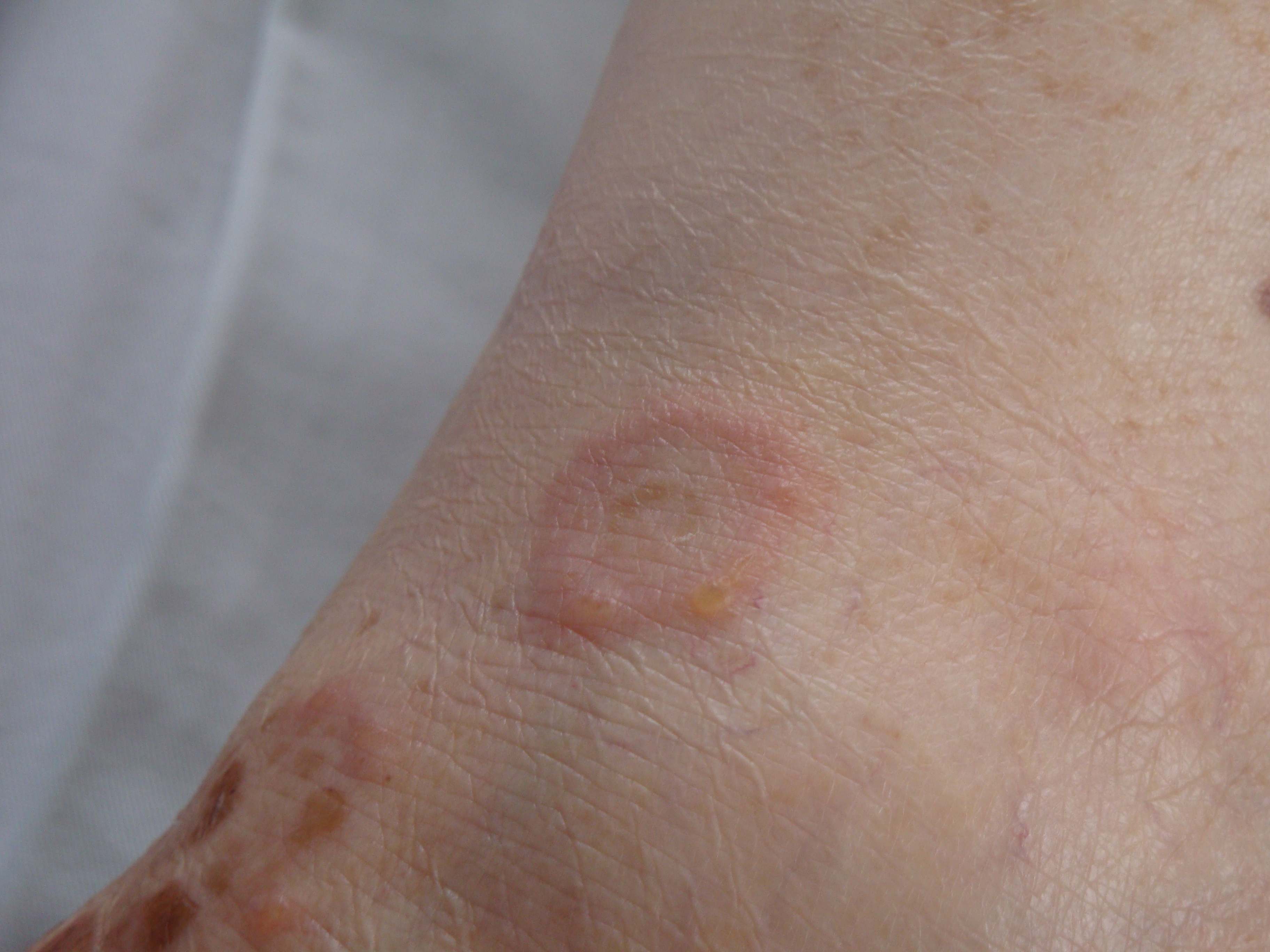
Pemphigus Herpetiformis. This image shows a well-demarcated ring-shaped lesion with clustered vesicles and surrounding erythema, consistent with pemphigus herpetiformis.
Contributed by Talel Badri (2013)
References
Daniel BS, Murrell DF. Review of autoimmune blistering diseases: the Pemphigoid diseases. Journal of the European Academy of Dermatology and Venereology : JEADV. 2019 Sep:33(9):1685-1694. doi: 10.1111/jdv.15679. Epub 2019 Jul 11 [PubMed PMID: 31087464]
Shimizu Y, Wakabayashi K, Hayashi Y, Hara K, Aoyama R, Niimi T, Tomino Y, Wada R, Hata M, Suzuki Y. MPGN Type 3 Associated with Pemphigus Herpetiformis Mimicking PGNMID and Dermatitis Herpetiformis. Case reports in nephrology and dialysis. 2019 Jan-Apr:9(1):15-24. doi: 10.1159/000498939. Epub 2019 Mar 21 [PubMed PMID: 31019928]
Level 3 (low-level) evidenceKiritsi D, Schauer F. [Autoimmune blistering dermatoses in children]. Der Hautarzt; Zeitschrift fur Dermatologie, Venerologie, und verwandte Gebiete. 2019 Apr:70(4):277-282. doi: 10.1007/s00105-019-4379-7. Epub [PubMed PMID: 30941468]
Witte M, Zillikens D, Schmidt E. Diagnosis of Autoimmune Blistering Diseases. Frontiers in medicine. 2018:5():296. doi: 10.3389/fmed.2018.00296. Epub 2018 Nov 2 [PubMed PMID: 30450358]
Kridin K, Schmidt E. Epidemiology of Pemphigus. JID innovations : skin science from molecules to population health. 2021 Mar:1(1):100004. doi: 10.1016/j.xjidi.2021.100004. Epub 2021 Feb 20 [PubMed PMID: 34909708]
Costa LMC, Cappel MA, Keeling JH. Clinical, pathologic, and immunologic features of pemphigus herpetiformis: a literature review and proposed diagnostic criteria. International journal of dermatology. 2019 Sep:58(9):997-1007. doi: 10.1111/ijd.14395. Epub 2019 Mar 22 [PubMed PMID: 30900757]
Hofmann SC, Juratli HA, Eming R. Bullous autoimmune dermatoses. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG. 2018 Nov:16(11):1339-1358. doi: 10.1111/ddg.13688. Epub [PubMed PMID: 30395395]
van Beek N, Zillikens D, Schmidt E. Diagnosis of autoimmune bullous diseases. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG. 2018 Sep:16(9):1077-1091. doi: 10.1111/ddg.13637. Epub [PubMed PMID: 30179336]
Jerbi A, Hachicha H, Feki S, Bahloul E, Sellami K, Abida O, Charfi S, Bouzid A, Sellami Boudawara T, Turki H, Masmoudi A, Masmoudi H. Pemphigus herpetiformis in South Tunisia: a clinical expression of pemphigus foliaceus? International journal of dermatology. 2018 Sep:57(9):1094-1101. doi: 10.1111/ijd.14139. Epub 2018 Jul 16 [PubMed PMID: 30011065]
Kakurai M, Takeshita H, Koga H, Ishii N, Moriyama Y. A Case of Pemphigus Herpetiformis Strongly Associated with Gastric Cancer. Acta dermato-venereologica. 2024 Dec 5:104():adv41319. doi: 10.2340/actadv.v104.41319. Epub 2024 Dec 5 [PubMed PMID: 39636071]
Level 3 (low-level) evidenceLakoš Jukić I, Jerković Gulin S, Marinović B. Blistering diseases in the mature patient. Clinics in dermatology. 2018 Mar-Apr:36(2):231-238. doi: 10.1016/j.clindermatol.2017.10.014. Epub 2017 Oct 3 [PubMed PMID: 29566927]
Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunologic research. 2018 Apr:66(2):255-270. doi: 10.1007/s12026-018-8986-7. Epub [PubMed PMID: 29479654]
Level 3 (low-level) evidenceKühn J, Giner T, Benoit S, Goebeler M, Wobser M. Pemphigus foliaceus transforming into pemphigus herpetiformis. Journal der Deutschen Dermatologischen Gesellschaft = Journal of the German Society of Dermatology : JDDG. 2023 Jun:21(6):655-657. doi: 10.1111/ddg.15037. Epub 2023 May 1 [PubMed PMID: 37125493]
Van Rhijn BD, Van Der Schaft J, Horvath B, Van Dijk MR, Pas HH. Dermpath & Clinic: Pemphigus herpetiformis with vacuolar interface dermatitis and autoantibodies against desmoglein 1 and 3. European journal of dermatology : EJD. 2021 Jun 1:31(3):433-435. doi: 10.1684/ejd.2021.4075. Epub [PubMed PMID: 34309538]
Kavita R, Sheenam A, Neelima B, Jitender BS. Unusual direct immunofluorescence (DIF) pattern in pemphigus herpetiformis- A case report. Indian journal of pathology & microbiology. 2023 Oct-Dec:66(4):852-854. doi: 10.4103/IJPM.IJPM_879_20. Epub [PubMed PMID: 38084547]
Level 3 (low-level) evidenceYamamoto M, Kokubu H, Yamamoto B, Koga H, Ishii N, Fujimoto N. Dipeptidyl peptidase-4 inhibitor-related pemphigus herpetiformis with antibodies against desmocollin 3. The Journal of dermatology. 2024 Jan:51(1):e16-e18. doi: 10.1111/1346-8138.16956. Epub 2023 Sep 7 [PubMed PMID: 37680081]
Sánchez-Pérez AP, Urbina-Calderón F, Pretell-Vera J, Principe-Pereda K, Saldaña-Beltrán C, Raza-Calderón R, Revilla-Torres A, Valverde-López J. A case of pemphigus herpetiformis with excellent response to mycophenolate mofetil. Acta dermatovenerologica Alpina, Pannonica, et Adriatica. 2021 Jun:30(2):87-88 [PubMed PMID: 34169707]
Level 3 (low-level) evidenceMarniquet ME, Joly P, Dansette D, Fenot M. A case of pemphigus herpetiformis successfully treated with rituximab. Journal of the European Academy of Dermatology and Venereology : JEADV. 2022 Sep:36(9):e686-e688. doi: 10.1111/jdv.18122. Epub 2022 Apr 8 [PubMed PMID: 35363902]
Level 3 (low-level) evidenceHamed M, Krausz J, Ziv M, Barak EC. Pediatric pemphigus herpetiformis treated with rituximab. JAAD case reports. 2024 Dec:54():98-104. doi: 10.1016/j.jdcr.2024.09.009. Epub 2024 Oct 28 [PubMed PMID: 39687069]
Level 3 (low-level) evidencePeterman CM, Vadeboncoeur S, Schmidt BA, Gellis SE. Pediatric Pemphigus Herpetiformis: Case Report and Review of the Literature. Pediatric dermatology. 2017 May:34(3):342-346. doi: 10.1111/pde.13152. Epub [PubMed PMID: 28523900]
Level 3 (low-level) evidence