Introduction
Cat eye syndrome (CES), also known as Schmid-Fraccaro syndrome, is a rare genetic disorder named for the vertical iris coloboma observed in some affected individuals. The condition is classically characterized by a triad of features—iris coloboma, anal atresia, and preauricular pits or tags.[1] However, CES can also involve a range of abnormalities affecting the neurodevelopmental, ocular, auricular, nasal, cardiovascular, gastrointestinal, and urogenital systems (see Image. Schematic Diagram Showing Iris Coloboma in Cat Eye Syndrome).[1]
The clinical presentation of CES is variable, with differences in affected organ systems, prognosis, genetics, and heritability among individuals. CES is a rare chromosomal disorder first described in the early 1960s by Schmid and Fraccaro. This condition is characterized by a partial tetrasomy or trisomy of chromosome 22q11.1-q11.2. The name "cat eye" originates from ocular colobomas—iris defects—present in about half of affected individuals, which give the pupil a distinctive keyhole or cat-eye appearance. Although ocular coloboma is the eponymous hallmark, CES is fundamentally a multisystem genomic disorder with highly variable expressivity, spanning a spectrum from nearly asymptomatic to severe anomalies across ocular, cardiac, renal, gastrointestinal, skeletal, and neurodevelopmental domains.
The association between ocular coloboma and anal atresia was first described by Haab in 1878.[1] The genetic alteration is due to a small supernumerary marker chromosome (sSMC), which was first described in 1965.[1] Schachenmann et al reported 3 pediatric patients and 1 patient’s mother who carried an additional, abnormally small chromosome featuring a submedian centromere, while the rest of the karyotype appeared normal.[1] This sSMC contains the CES critical region (CESCR), located within the proximal portion of chromosome 22q11.2, between the centromere and the LCR22-A region.[2] Additional genetic conditions related to chromosome 22 include the oculo-auriculo-vertebral spectrum (OAVS), DiGeorge syndrome, and mosaic trisomy 22.
At the genetic level, CES arises from a supernumerary marker chromosome, often dicentric, composed of material from chromosome 22. In approximately 90% of cases, this marker contains 2 extra copies of the proximal 22q11 region (tetrasomy), while a smaller proportion exhibits an additional copy (trisomy). The critical region encompasses approximately 1.5 to 2 Mb and includes multiple dosage-sensitive genes whose overexpression is believed to contribute to the diverse phenotypic features of CES. Molecular cytogenetic techniques, such as fluorescence in situ hybridization (FISH), array comparative genomic hybridization (aCGH), and, more recently, genome-wide single-nucleotide polymorphism (SNP) microarrays, have replaced traditional karyotyping for precise delineation of the supernumerary chromosome and identification of the breakpoints. This high-resolution genomic mapping is crucial for definitive diagnosis, genotype-phenotype correlations, and recurrence-risk counseling.[3]
Clinically, CES is remarkably heterogeneous. The classic triad comprises iris coloboma, preauricular skin tags or pits, and anal atresia or other anorectal malformations. However, no single feature is universally present. Iris coloboma appears in 40% to 60% of cases, preauricular anomalies in up to 70%, and anorectal malformations in about 30% to 50%. Cardiac defects, most commonly total or partial atrioventricular septal defects and tetralogy of Fallot, occur in approximately half of patients and are a major contributor to early morbidity and mortality. Renal anomalies, reported in 20% to 40% of cases, may include unilateral renal agenesis, duplex collecting systems, hydronephrosis, and vesicoureteral reflux. Skeletal abnormalities range from vertebral segmentation defects to limb anomalies. Otolaryngological manifestations may include hearing loss from middle-ear dysplasia. Less commonly, gastrointestinal anomalies beyond anorectal malformations—such as duodenal atresia or Hirschsprung disease—have also been documented.[4]
Neurodevelopmental outcomes in CES vary widely. Although some children achieve developmental milestones within normal limits, others present with global developmental delay, intellectual disability, or features consistent with autism spectrum disorder. Hypotonia during infancy and feeding difficulties—often related to underlying gastrointestinal anomalies—may further compromise early growth. Growth parameters can be affected, with some patients exhibiting short stature or failure to thrive; however, many ultimately achieve normal height and weight. Behavioral phenotypes—such as attention-deficit/hyperactivity disorder (ADHD) and anxiety disorders—have also been reported, emphasizing the importance of comprehensive developmental and psychological assessment.[5]
Ophthalmic manifestations in CES extend beyond the iris coloboma. Additional anomalies such as chorioretinal colobomas, microphthalmia, cataracts, microcornea, and strabismus can contribute to visual impairment. A comprehensive ophthalmologic evaluation includes slit-lamp biomicroscopy to assess anterior segment abnormalities, indirect ophthalmoscopy for posterior segment examination, and optical coherence tomography (OCT), when available, to delineate the extent of colobomatous defects. Early detection and management of refractive errors, amblyopia, and strabismus are essential to support optimal visual development. Surgical intervention for coloboma is rarely indicated and is typically reserved for cases involving severe aniridia-like photophobia or significant cosmetic concerns (see Image. Schematic Diagram Showing Chorioretinal Coloboma in Cat Eye Syndrome).[6]
The management of CES depends on the organ systems involved and the severity of associated malformations. Given the significant clinical heterogeneity, an individualized, interprofessional approach is essential. This activity outlines the genetic and phenotypic spectrum of CES and outlines strategies for tailoring medical care to each patient’s needs. Cardiac evaluation at diagnosis is mandatory. Echocardiography within the first weeks of life is essential for detecting structural heart disease; in moderate-to-severe cases, surgical repair during infancy may be lifesaving. Long-term cardiology follow-up is critical to monitor for residual defects, arrhythmias, and pulmonary hypertension. Similarly, early renal ultrasonography is recommended to identify anatomical anomalies, guide urologic management, and prevent complications such as hypertension or renal insufficiency.[7]
Gastroenterological and colorectal management primarily focuses on anorectal malformations. Posterior sagittal anorectoplasty (PSARP) is the standard repair for imperforate anus, with timing and technical details tailored to the patient’s specific anatomy and overall health. Nutritional support—ranging from gavage or gastrostomy feeding in neonates to dietary modifications in older children—is essential, especially when gastrointestinal motility disorders or malabsorption are present.[8]
Audiologic and otologic care begins with newborn hearing screening. Conductive hearing loss due to middle ear anomalies may require interventions such as tympanostomy tubes or myringotomy. Speech therapy and educational support, tailored to the child’s developmental needs, are essential for optimizing communication outcomes. Genetic counseling for families includes discussion of recurrence risk, which is generally low (<1%) in de novo cases but higher in familial instances when a parent carries the small supernumerary marker chromosome in a balanced form.[6]
Secondary complications may include endocrine disorders, particularly growth hormone deficiency and thyroid dysfunction, necessitating regular endocrinologic screening. Orthopedic evaluations focus on detecting scoliosis and limb-length discrepancies. Dental and orthodontic assessments help identify malocclusion and enamel hypoplasia. Psychosocial support for families—including referrals to patient advocacy groups and peer support networks—promotes coping strategies and shared experiences.[9]
From a research perspective, CES provides valuable insights into gene dosage effects in contiguous-gene syndromes. The 22q11 region implicated in CES overlaps with that of DiGeorge syndrome (22q11.2 deletion), yet their phenotypes differ, reflecting divergent consequences of haploinsufficiency versus gene overexpression. Current studies focus on elucidating the roles of candidate genes such as CECR1 (which encodes adenosine deaminase 2) and CECR2 (involved in chromatin remodeling) in contributing to CES manifestations. Animal models with targeted duplications of the 22q11 region are under development to investigate relevant developmental pathways. Additionally, next-generation sequencing techniques show promise in detecting cryptic rearrangements and refining genotype–phenotype correlations, ultimately improving prognostic accuracy and identifying potential therapeutic targets.[10]
In summary, CES is a complex, multisystem chromosomal disorder. While its hallmark features include ocular coloboma, ear anomalies, and anorectal malformations, the condition also encompasses a wider phenotypic spectrum affecting the cardiac, renal, skeletal, neurodevelopmental, and endocrine systems. Accurate diagnosis relies on high-resolution cytogenetic and molecular techniques. Effective management requires coordinated multidisciplinary care, involving specialties from neonatology and cardiology to ophthalmology, urology, and developmental pediatrics. As advances in molecular genetics continue, they will enhance personalized prognostic counseling and enable the development of targeted therapies, ultimately improving outcomes for individuals and families affected by this rare but informative genomic syndrome.[3]
Etiology
Register For Free And Read The Full Article
 Search engine and full access to all medical articles
Search engine and full access to all medical articles- 10 free questions in your specialty
- Free CME/CE Activities
- Free daily question in your email
- Save favorite articles to your dashboard
- Emails offering discounts
Learn more about a Subscription to StatPearls Point-of-Care
Etiology
The etiology of CES results from a heterogeneous group of genetic alterations, all of which involve extrachromosomal material containing the CESCR located on chromosome 22. The following genetic alterations have been reported to be associated with CES:
- Type 1 sSMC, containing only the CESCR [4]
- Type 2 sSMC, containing both the CESCR and DiGeorge syndrome critical region [11]
- Intrachromosomal duplication of the CESCR [12]
- Intrachromosomal triplication of the CESCR [13]
- Unknown genetic alterations
Both CES and DiGeorge syndrome involve genetic alterations at the chromosome region 22q11.2. CES is caused by a duplication of this region, often manifested as a small supernumerary marker chromosome, whereas DiGeorge syndrome results from a microdeletion. In CES, the formation of the small supernumerary marker chromosome typically occurs through an inverted duplication of chromosome 22, believed to arise from spontaneous recombination during meiosis, with most cases occurring de novo.[14] Rarely, CES may be inherited due to meiotic nondisjunction in parents carrying a balanced translocation [15] or from a parent with mosaicism.[4] No environmental factors have been identified as contributing to the development of CES.
CES arises from a chromosomal imbalance involving the proximal region of chromosome 22 (22q11.1-q11.2). In over 90% of affected individuals, this manifests as a supernumerary marker chromosome—often dicentric—harboring 2 additional copies of an approximately 1.5 to 2 Mb critical region, resulting in partial tetrasomy of 22q11.1-q11.2. A minority of cases exhibit a similarly derived marker with only an additional copy, leading to partial trisomy. This marker chromosome typically includes dosage-sensitive genes whose overexpression disrupts normal embryonic development across multiple organ systems.[10]
Chromosomal Mechanisms
- De novo formation: The vast majority of supernumerary marker chromosomes in CES arise spontaneously during gametogenesis or early embryogenesis. Breakpoints cluster within low-copy-repeat (LCR) regions flanking 22q11, predisposing to nonallelic homologous recombination and the formation of small extra chromosomes.
- Mosaicism: Approximately 20 % of patients exhibit mosaicism, with some cells carrying the marker chromosome while others remain disomic. Mosaic individuals often present with milder or more variable phenotypes.
- Familial transmission: In rare cases (<5%), a parent carries the small marker chromosome in a balanced form (often mosaic), conferring a recurrence risk up to 50% for offspring.[16]
Dosage-Sensitive Candidate Genes
Several genes within the duplicated critical region have been implicated in the pathogenesis of CES:
- CECR1 (ADA2): This gene encodes adenosine deaminase 2; dysregulated activity may contribute to vascular abnormalities and immune dysfunction.
- CECR2: This gene functions as a chromatin remodeling factor; overexpression may disrupt the development of neural crest–derived tissues.
- SLC7A4: This gene encodes a cationic amino acid transporter; potentially involved in renal and gastrointestinal morphogenesis.
- TXNRD2: This gene encodes a mitochondrial thioredoxin reductase; dosage changes may affect cellular oxidative balance.[16]
Genotype-Phenotype Correlations
Although no strict correlation exists between marker size and clinical severity, larger markers that include additional 22q11 material tend to cause more pronounced malformations. Mosaicism typically results in a milder phenotype. The presence of 2 intact centromeres may increase the mitotic stability of the marker chromosome, often leading to uniform tetrasomy across multiple tissues.[17]
Table 1. Marker Types and Phenotypic Implications in Cat Eye Syndrome
|
Marker Type |
Chromosome Content |
Frequency |
Phenotypic Implication |
|
Type I (inv dup) |
Inverse duplication(22)(q11.1) |
~70 % |
Classic CES features with variable expressivity |
|
Type II (min marker) |
Small supernumerary marker chromosome |
~20 % |
Often mosaic, milder, or atypical |
|
Partial trisomy |
Single extra 22q11 segment |
~10 % |
Milder dosage effects; overlaps with tetrasomy phenotype |
Inheritance Patterns and Recurrence Risk
Most CES cases occur sporadically, with parental carriers being rare. However, when mosaicism is present in a parent, cytogenetic evaluation is essential, as the recurrence risk for future pregnancies may be as high as 50% when detected. Prenatal diagnosis through amniocentesis or chorionic villus sampling, combined with FISH or microarray analysis, can identify both de novo and inherited marker chromosomes.[18]
In summary, CES arises from dosage alterations of critical developmental genes on 22q11.1 and 22q11.2 caused by a small supernumerary marker chromosome. The combination of de novo marker formation, possible mosaicism, and rare familial transmission contributes to the broad phenotypic spectrum—from isolated ocular colobomas to complex multisystem malformations—that defines this syndrome.
Epidemiology
CES has an estimated incidence of approximately 1 in 150,000 live births.[19] Over 300 cases have been reported in the medical literature, although many individuals with mild symptoms likely remain undiagnosed. Both males and females are affected equally. The genetic etiology is an sSMC in 91% of cases, mosaicism was found in 40% of cases, and parental transmission of an sSMC was seen in 23% of cases.[4]
CES is a rare chromosomal disorder characterized by partial tetrasomy or trisomy of chromosome 22q11.1-q11.2, with an estimated incidence of approximately 1 in 50,000 to 1 in 100,000 live births. Due to wide phenotypic variability—from nearly asymptomatic individuals to those with multiple congenital anomalies—the actual prevalence may be higher, as milder cases can often go undiagnosed or are misattributed to isolated ocular or anorectal malformations. Both males and females are affected equally, with no known predilection for any particular ethnic or geographic populations.[3]
The vast majority of CES cases arise de novo, with no prior family history, resulting from sporadic nonallelic homologous recombination events within LCR regions of chromosome 22 during gametogenesis. However, familial transmission accounts for approximately 5% to 10% of cases, typically involving a parent who carries the small supernumerary marker chromosome, often in mosaic form. In such cases, the recurrence risk for future pregnancies may approach 50%, highlighting the importance of parental cytogenetic analysis and comprehensive genetic counseling following a CES diagnosis.[4]
Mosaicism for the marker chromosome occurs in approximately 15% to 25% of individuals with CES and is typically associated with milder or more variable clinical manifestations. In mosaic CES, only a subset of cells contains the supernumerary chromosomal material, which attenuates the gene dosage effect and often results in a less severe phenotype. Detecting mosaicism requires high-resolution molecular cytogenetic techniques, such as FISH or chromosomal microarray analysis, as conventional karyotyping of peripheral blood lymphocytes may fail to identify low-level mosaicism present in other tissues.[20]
Clinically recognized cases of CES typically present in infancy or early childhood due to prominent congenital anomalies. Anal atresia or imperforate anus, observed in 30% to 50% of patients, often prompts neonatal evaluation and facilitates early cytogenetic testing. Ocular colobomas, present in 40% to 60% of cases, and preauricular skin tags or pits, seen in up to 70%, are similarly distinctive features that frequently trigger genetic workup. Cardiac defects are identified in approximately 50% of individuals, with atrioventricular septal defects and tetralogy of Fallot among the most common. These cardiac lesions may contribute to early infant mortality if not promptly addressed but can also be detected prenatally through fetal echocardiography—particularly when 22q11 duplications are identified on prenatal chromosomal microarray.[5]
Renal malformations occur in 20% to 40% of individuals with CES, including unilateral renal agenesis, duplex collecting systems, and vesicoureteral reflux. Routine renal ultrasonography in affected infants can detect these often asymptomatic anomalies, allowing for timely urologic intervention. Growth patterns in CES are variable—some children experience failure to thrive during infancy—often due to associated gastrointestinal anomalies—whereas others attain normal height and weight percentiles by school age. Endocrine dysfunctions, including thyroid abnormalities and growth hormone deficiency, have been reported but remain poorly characterized epidemiologically.[4]
Neurodevelopmental outcomes in CES vary widely. Approximately 30% to 50% of affected children exhibit global developmental delay or intellectual disability, while a smaller subset presents with specific learning disabilities or behavioral disorders, including ADHD and autism spectrum features. Due to this variability, formal developmental screening and early intervention are recommended for all patients. Epidemiological studies suggest that such developmental support improves long-term educational and psychosocial outcomes.[6]
Advances in prenatal screening—particularly noninvasive prenatal testing (NIPT) and high-resolution chromosomal microarray—have improved the early detection of 22q11 duplications, sometimes even before sonographic anomalies are apparent. However, the positive predictive value of NIPT for small supernumerary marker chromosomes is lower than that for more common aneuploidies, necessitating confirmatory invasive testing. Reported series indicate that up to 20% of prenatal diagnoses of 22q11.2 duplications result in adverse pregnancy outcomes, including miscarriage or termination, often influenced by the detection of cardiac or severe renal anomalies on fetal imaging.[7]
In summary, CES is a rare chromosomal disorder with an estimated incidence of 1 in 50,000 to 1 in 100,000 live births, affecting males and females equally. Although the majority of cases arise de novo, familial transmission and mosaicism account for a minority of presentations and contribute to the condition’s phenotypic variability. Key diagnostic indicators—such as anal atresia, ocular colobomas, preauricular tags, and congenital heart defects (CHD)—often prompt early cytogenetic evaluation. Although advances in prenatal screening have increased detection rates, the complete epidemiological profile remains unclear, particularly for individuals with mild or subclinical manifestations. Ongoing refinement of molecular diagnostics and population-based studies will be critical to determining true prevalence, clarifying recurrence risks, and guiding long-term clinical surveillance strategies for this multisystem genomic syndrome.[8]
Pathophysiology
The exact pathophysiology of CES remains incompletely understood but centers on the gene products within the CESCR. Fundamentally, CES arises from a gene dosage imbalance on the proximal segment of chromosome 22 (22q11.1-q11.2). In most cases, a small supernumerary marker chromosome duplicates this critical region, resulting in partial trisomy or tetrasomy and increasing the copy number of multiple contiguous, dosage-sensitive genes. During early embryogenesis, this gene overexpression perturbs developmental pathways across several germ layers, leading to the characteristic multisystem malformations of CES.[8]
Neural Crest–Derived Structures
Many anomalies in CES affect tissues derived from the neural crest. The periocular mesenchyme, originating from cranial neural crest cells, forms the iris stroma and eyelid connective tissue. Dysregulation of CECR2 and other chromatin-remodeling genes within the duplicated segment disrupts normal migration and differentiation of these cells, leading to iris colobomas and epibulbar dermoids. Similarly, aberrant neural crest contributions to the first and second pharyngeal arches result in preauricular skin tags, auricular malformations, and defects in pharyngeal pouch derivatives that form the thymus and parathyroids observed in other 22q11 syndromes.[21]
Cardiovascular Development
The endothelial-to-mesenchymal transition that drives endocardial cushion formation in the atrioventricular canal is highly sensitive to gene dosage within 22q11.1-q11.2. Elevated CECR1 (ADA2) activity may disrupt local adenosine gradients essential for proper cushion maturation, while overexpression of genes adjacent to TBX1 interferes with neural crest–derived contributions to the cardiac outflow tract. Consequently, CES is associated with a high incidence of atrioventricular septal defects, tetralogy of Fallot, and interrupted aortic arch.[9]
Gastrointestinal and Genitourinary Morphogenesis
Precise coordination between the hindgut endoderm and adjacent mesenchyme is essential for normal anorectal and renal development. Overexpression of SLC7A4 and other solute transporter genes may disrupt ion homeostasis in the developing cloaca, contributing to anorectal malformations—such as anal atresia, stenosis, or fistulae—in up to 50% of cases. Concurrently, excessive TXNRD2-mediated alterations in redox balance can impair mesonephric duct signaling to the metanephric blastema, leading to renal anomalies including agenesis, dysplasia, or ureteral duplication.[16]
Neurodevelopment and Growth
In addition to structural anomalies, gene dosage imbalances disrupt neurogenesis and synaptic maturation, contributing to the variable neurodevelopmental phenotypes observed in CES. Overexpression of CECR2 in the developing forebrain may impair neuronal migration and cortical lamination, which can underlie intellectual disability and autism spectrum features in some patients. Additionally, altered growth factor signaling—potentially through dysregulation of genes such as LZTR1 within the duplicated region—may result in short stature and feeding difficulties, often exacerbated by associated gastrointestinal anomalies.[7]
Mosaicism and Phenotypic Variability
Approximately 20% of individuals with CES exhibit mosaicism, where only a subset of cells carry the extra chromosome. This cellular mosaicism reduces the overall gene dosage effect, often resulting in milder or atypical phenotypes, such as isolated ocular coloboma or subtle preauricular tags in otherwise healthy adults. The proportion of marker-positive cells in key tissues influences the severity and specific combination of malformations in each case.[18]
In summary, CES arises from copy-number–driven overexpression of multiple contiguous genes on chromosome 22q11.1-q11.2, which disrupts key developmental processes such as neural crest migration, endocardial cushion formation, hindgut morphogenesis, and neurodevelopment. The interplay of gene dosage, tissue-specific sensitivity, and mosaicism contributes to the broad phenotypic spectrum, including ocular, cardiac, gastrointestinal, renal, and neurodevelopmental anomalies, that characterizes this multisystem genomic disorder.[22]
Histopathology
CES is characterized by the presence of a small supernumerary marker chromosome involving the 22q11.1-q11.2 region, and its histopathological features reflect disrupted embryological development across multiple tissues. In ocular specimens, colobomas typically demonstrate focal absence of the iris stroma and anterior uveal tissue, with the defect often lined by neuroectodermal pigmented epithelium continuous with the posterior iris pigment layer.
Light microscopy reveals a sharply demarcated gap in the anterior segment, with attenuated or absent sphincter and dilator muscles at the margins of the coloboma. The adjacent stroma may show reactive fibrovascular proliferation and occasional melanin-laden macrophages, indicative of chronic tissue remodeling. In chorioretinal colobomas, the retina and choroid terminate abruptly at the lesion’s edge, where retinal pigment epithelium is preserved but overlying photoreceptor and neural layers are absent.[20]
Preauricular skin tags and pits, when examined histologically from excised tissue, reveal rudimentary derivatives of the branchial arches. Histologically, these tags consist of stratified squamous epithelium overlying a core of loose fibrovascular stroma, often containing adnexal structures such as eccrine glands and hair follicles. In some specimens, minor cartilage nodules or neural crest–derived mesenchyme are present, reflecting dysgenesis of the first and second pharyngeal arches.
Anal atresia specimens—typically resected during PSARP—demonstrate a blind-ending colorectal pouch lined by colonic-type mucosa that abruptly transitions to stratified squamous epithelium without an intervening anal transition zone. The muscularis propria is frequently disorganized, with incomplete development of the inner circular and outer longitudinal muscle layers. The lamina propria may show variable inflammatory infiltrates, particularly in the setting of fecal stasis or microperforation.[23]
Cardiac histology—available from rare autopsy or explant specimens—demonstrates characteristic features of endocardial cushion defects. Atrioventricular septal defects typically exhibit deficient or absent mesenchymal tissue in the superior and inferior cushions, resulting in a common atrioventricular orifice and intermingling of valve leaflets. Immunohistochemical staining for neural crest cell markers (eg, Sox10) reveals reduced or aberrant distribution within the cushion mesenchyme. In the great vessels, tetralogy of Fallot specimens often exhibit anterior malalignment of the conal septum and right ventricular outflow tract obstruction, accompanied by marked right ventricular myocardial hypertrophy.[24]
Renal histopathology in CES spans a spectrum from unilateral renal agenesis, characterized by complete absence of renal tissue on one side, to dysplastic kidneys characterized by disorganized glomeruli, primitive tubules embedded in fibrous stroma, and occasionally dilated collecting ducts. In duplex systems, the presence of 2 ureteric buds gives rise to separate pelvicalyceal systems, with histology revealing duplicated urothelial-lined lumens and lamina propria.[19]
At the cellular level, affected tissues consistently exhibit aberrant neural crest migration and mesenchymal differentiation, reflecting overexpression of genes such as CECR2 and CECR1 within the duplicated 22q11 region. Electron microscopy—performed in isolated research cases—demonstrates disorganized basement membranes at the coloboma margins and altered extracellular matrix composition in cardiac cushions. Together, histopathological findings across ocular, cutaneous, anorectal, cardiac, and renal specimens underscore the multisystem nature of CES, revealing the anatomic substrates of its clinical features and providing insight into the developmental pathways perturbed by chromosomal dosage imbalance.[20]
Toxicokinetics
Table 2. Pharmacokinetics of Commonly Used Agents in the Management of Cat Eye Syndrome
|
Drug |
Indication |
Absorption and Distribution |
Metabolism |
Elimination |
Clinical Notes |
|
Propofol |
General anesthesia (neonatal surgery) |
Rapid intravenous distribution; Vd ~4-10 L/kg; high brain penetration |
Hepatic conjugation by UDP-glucuronosyltransferases (UGTs) |
Renal excretion of metabolites |
Reduced clearance in neonates → lower infusion rates; monitor hemodynamics |
|
Sevoflurane |
Inhalational anesthesia |
Rapid alveolar uptake; Fa/Fi ~0.6; highly perfusion-dependent |
Minimal hepatic metabolism (~5% by CYP2E1) |
Primarily exhaled; <3% renal metabolite excretion |
Low blood solubility → fast induction/emergence; monitoring for nephrotoxic hexamethyldisilazane (HMDS) metabolite |
|
Furosemide |
Heart failure, pulmonary edema |
Variable oral bioavailability (50–90%); extensive tissue binding |
Minor hepatic glucuronidation |
Renal excretion of the unchanged drug |
CES-related cardiac defects may alter Vd and clearance; monitoring for electrolytes closely |
|
Digoxin |
Inotrope in congenital heart disease |
Oral absorption ~70%; distributes to myocardium (Vd ~7 L/kg) |
Minimal hepatic metabolism |
Renal excretion; unchanged (~60-80%) |
Narrow therapeutic index; impaired renal function in CES warrants dosage adjustment |
Gene-Dosage–Driven Metabolic Pathway Alterations
The tetrasomy of 22q11 in CES involves duplication of genes encoding metabolic enzymes and transporters, resulting in subtle alterations in endogenous substrate processing.
Table 3. Impact of 22q11 Gene Overexpression on Metabolic Functions and Clinical Outcomes
|
Gene |
Normal Function |
Effect of Overexpression |
Clinical Implications |
|
ADA2 |
Catalyzes the deamination of adenosine to inosine |
↑ ADA2 activity → reduced adenosine levels |
May alter immunomodulation, vascular tone, and potentially influence drug–receptor interactions |
|
TXNRD2 |
Mitochondrial thioredoxin reductase |
↑ TXNRD2 activity → altered redox balance |
Modulates oxidative stress in hepatocytes → potential impact on drug metabolism rates |
|
SLC7A4 |
Mediates cationic amino acid transporter |
↑ Transporter expression → altered arginine uptake |
Impacts nitric oxide synthesis and may modify first-pass drug absorption in enterocytes |
Integrating Pharmacogenomics and Therapeutic Monitoring
- Propofol clearance: Neonatal UDP-glucuronosyltransferase (UGT) activity is immature and may be further modulated by oxidative stress pathways altered in CES. This can reduce propofol clearance, warranting lower infusion rates and close blood pressure monitoring.[25]
- Inhalational agents: Although sevoflurane undergoes minimal hepatic metabolism, shifts in redox homeostasis—potentially driven by TXNRD2 overexpression—may affect CYP2E1 activity. Anesthesiologists should remain alert for delayed emergence.[26]
- Renal-excreted drugs: Dosing of furosemide and digoxin in CES should consider the potential for renal dysgenesis. Baseline renal ultrasound and glomerular filtration rate (GFR) assessment are essential for guiding initial dosing and ongoing therapeutic drug monitoring.[27]
Clinical Implications
Recognizing the impact of CES-related gene dosage on pharmacokinetics is crucial for safe anesthesia and cardiac care. Personalized dosing—guided by altered enzyme activity, organ function evaluation, and therapeutic drug monitoring—enhances treatment efficacy while reducing toxicity in this vulnerable population.
History and Physical
The phenotypic spectrum of CES is highly variable. Patient history often includes congenital anomalies identified at birth. Although most cases are sporadic, rare familial occurrences have been reported. In these instances, the mosaic parent carries an sSMC with a low percentage of affected cells and exhibits minimal or no clinical features.[4] A comprehensive physical examination is critical, as findings are heterogeneous and usually involve multiple organ systems.
Preauricular skin tags or pits are the most common clinical finding, and they are present in approximately 81% of patients.[28] However, only 16% display the full “classic triad” of iris coloboma, anal atresia, and preauricular anomalies, while 9% exhibit none of these classic signs.[28] Cardiac anomalies are reported in 51% of cases, followed by intellectual disability (47%), ocular motility disturbances (45%), gastrointestinal malformations (44%), ophthalmologic anomalies (35%), and genitourinary tract defects (32%).[4] These clinical findings of CES can be systematically categorized according to the organ systems involved.
Patient History
Prenatal and birth history
- Pregnancy is often uneventful, though polyhydramnios or intrauterine growth restriction may be noted on prenatal ultrasound.
- Delivery typically occurs at term or late preterm, with birth weight often low for gestational age.
- Early feeding difficulties may arise due to anorectal malformations or generalized hypotonia.[29]
Neonatal course
- Newborns may present with anal atresia or stenosis, often necessitating prompt surgical intervention.
- Feeding intolerance or bilious vomiting can occur due to underlying intestinal anomalies such as malrotation.[17]
- Cyanotic spells or heart murmurs may be evident in the presence of CHDs, most commonly total anomalous pulmonary venous return (TAPVR) or tetralogy of Fallot.[4]
Family and genetic history
- Most cases are sporadic, although rare instances of inheritance from a parent with a small supernumerary marker chromosome have been reported.
- While there is no consistent Mendelian inheritance pattern, occasional vertical transmission may occur from a mildly affected mosaic parent.[3]
Developmental history
- Infancy is often characterized by hypotonia and delayed gross motor milestones such as sitting and walking.
- Cognitive development varies, with many individuals exhibiting mild-to-moderate learning difficulties.
- Speech delay is common, with expressive language typically more affected than receptive abilities.[6]
Ophthalmologic history
- Colobomatous iris, often described as a "keyhole" defect, is typically identified at birth or during early eye examinations.
- Patients may have a history of recurrent eye infections or frequent eye rubbing, particularly if the visual axis is involved; large colobomas can cause photophobia.
- Strabismus or poor visual tracking is also commonly reported.[30]
Physical Examination
General appearance
- Growth parameters are typically at or below the 10th percentile for weight and length.
- Mild facial dysmorphisms may be noted, including downward-slanting palpebral fissures and hypertelorism.
- Generalized hypotonia is common, often presenting as a "floppy" posture.[9]
Head and neck—Eyes
- Iris coloboma is common, typically presenting as an inferonasal notch or “keyhole” pupil.
- Microphthalmia or mild microcornea may be present.
- Chorioretinal coloboma can be detected on funduscopic examination.[10]
Ocular and orbital anomalies
Ocular motility defects, such as Duane syndrome or strabismus, occur in 45% of patients, while 35% exhibit other ophthalmologic abnormalities.[28]The ocular findings in CES are variable and do not always include the characteristic "cat eye" iris colobomas, which are present in about one-third of cases.[28] Ocular colobomas may affect the iris, choroid, and/or retina. Additional ocular findings may include aniridia, microphthalmia, cataracts, and corneal clouding. Orbital features often include hypertelorism and down-slanting palpebral fissures.
Ears
- Preauricular skin tags, pits anterior to the tragus, or fistulae are common.
- Some patients have low-set, malformed pinnae.
- Conductive hearing loss may result from external canal stenosis or middle ear anomalies.
- Prominent antihelices, hypoplastic earlobes, and auditory canal atresia are also associated with hearing impairment.
- Preauricular tags represent the most consistent feature of this syndrome.[21]
Mouth
- Cleft palate or high-arched palate occurs in up to 20% of cases.
- Micrognathia may also be present.[9]
Nasal malformations
Nasal malformations may include a broad and depressed nasal bridge and minimal epicanthal folds.[8]
Cardiovascular
- Heart murmurs, such as a systolic ejection murmur, may be present with a patent ductus arteriosus (PDA) and a ventricular septal defect (VSD).
- Signs of heart failure (eg, tachypnea and hepatomegaly) occur with significant structural defects.
- Although rare, arrhythmias can arise from conduction system anomalies.
- CHD affects approximately half of CES patients, making it the second most common clinical feature.
- Several types of CHDs have been associated with CES, including TAPVR, tetralogy of Fallot, tricuspid atresia, VSDs, and atrial septal defects (ASDs), as well as systemic venous anomalies such as persistent left superior vena cava and absence of the inferior vena cava.[28]
- The most commonly reported cardiac anomalies in CES are TAPVR, ASDs, and VSDs.[31] Notably, TAPVR, tetralogy of Fallot, and tricuspid atresia are classified as critical CHDs—life-threatening structural defects that can present with cyanosis and critical illness in the newborn period.
Abdomen and genitourinary
- Abdominal scars or stomas may be present from surgical repair of an imperforate anus.
- Palpable kidneys may indicate underlying renal anomalies, such as a horseshoe kidney, which is often detected on imaging.
- Hypospadias occurs in up to 25% of affected males.[10]
Gastrointestinal malformations
Gastrointestinal anomalies are present in approximately 44% of patients.[28] These may include anorectal malformations, anal atresia, biliary atresia, Hirschsprung disease, and intestinal malrotation. The most commonly reported anomaly is anal atresia (imperforate anus).
Urogenital malformations
Urogenital malformations are seen in 32% of patients.[28] Defects may include kidney hypoplasia or aplasia, ectopic kidney, bladder fistula, hydronephrosis, cryptorchidism, and hypospadias.
Extremities
- Mild limb asymmetry or clinodactyly may be observed.
- In some cases, digits may be hypoplastic or shortened.[30]
Neurologic
- Hypotonia with reduced deep tendon reflexes is common.
- Infants may exhibit poor head control.
- Coordination and gait abnormalities vary once the child becomes ambulatory.[8]
Skin
In addition to preauricular tags and pits, which are described above, patients may occasionally present with café-au-lait spots or haemangiomas.[6]
Abnormal neurodevelopmental outcomes and other miscellaneous anomalies
Neurodevelopmental and other associated anomalies may include intellectual disability, intrauterine growth restriction, short stature, congenital hip dysplasia, rib anomalies, and hypotonia. Intellectual disability occurs in fewer than half of the patients.[28] Hypothalamic-pituitary abnormalities leading to hormone deficiencies have been reported in approximately 22% of cases.[28]
Key Points
CES is characterized by the classic triad of ocular coloboma ("cat eye"), preauricular anomalies, and anorectal malformations, but it demonstrates significant phenotypic variability. Clinicians should conduct thorough screening for cardiac, renal, and craniofacial involvement to inform comprehensive multidisciplinary care.
Evaluation
Early evaluation and diagnosis are essential to prevent complications and improve outcomes. However, the wide spectrum of clinical phenotypes can make the evaluation and diagnosis of CES challenging. Patients may require assessment in the neonatal intensive care unit (NICU) depending on the severity of malformations, especially cardiac anomalies. The findings from a detailed physical examination will determine the choice of diagnostic tests, imaging, and subspecialty consultations.[3]
Subspecialist Consultations
Cardiology is the highest priority for assessing potentially critical CHD. In suspected CES cases, anticipating and evaluating cardiac malformations is essential, as these are the second most common feature reported, even though they are not part of the “classic triad.” Additional subspecialists who may be involved include ophthalmology, otolaryngology, gastroenterology, endocrinology, urology, neurology, general surgery, orthopedic surgery, cardiothoracic surgery, and genetics.[5]
Genetic Testing and Counseling
Karyotyping is typically sufficient for diagnosing CES, either prenatally or postnatally. However, FISH with specific probes may be necessary to detect mosaic forms of CES. In inherited cases, genetic counseling is recommended to help families assess the risk of recurrence and understand the implications of the diagnosis.[30]
Evaluation of Cat Eye Syndrome
Clinical history
- Prenatal and perinatal history: The clinician should review the prenatal course for signs such as polyhydramnios, intrauterine growth restriction, or ultrasound findings indicating potential cardiac or renal malformations.
- Family history: Although most cases of CES are de novo, the clinician should inquire about any history of similar ocular, anal, or auricular anomalies in family members.[3]
Physical examination
- Ocular: The clinician should evaluate for iris coloboma (the “cat-eye” defect), chorioretinal coloboma, microphthalmia, or microcornea. Vision, pupillary function, and extraocular movements should also be assessed.
- Auricular: Preauricular skin tags, sinuses, microtia, or anomalous ear cartilage should be inspected. Post-auricular pits should be palpated.
- Facial and cranial: Head circumference should be measured to assess for microcephaly or macrocephaly. Any facial asymmetry or hemifacial microsomia should be noted.
- Anal and genitourinary: The clinician should examine for imperforate anus, anal stenosis, or an anteriorly displaced anus. In male patients, evaluation should include assessment for hypospadias or cryptorchidism.
- Cardiovascular: Cardiac auscultation should be performed to detect murmurs, and the patient should be inspected for signs of cyanosis or poor perfusion.
- Renal: The abdomen should be palpated for flank masses that may indicate a horseshoe kidney.
- Growth and development: Growth parameters, including weight, length, and head circumference, should be recorded and compared with normative charts. The patient should be screened for developmental delay and hypotonia.[6]
Ophthalmic investigations
- Slit-lamp biomicroscopy: This technique is used to characterize the size and location of the iris coloboma and to assess anterior segment structures.
- Fundus examination (direct or indirect ophthalmoscopy): This is performed to evaluate for chorioretinal coloboma, optic nerve dysplasia, or macular involvement.
- Anterior segment optical coherence tomography: This procedure helps quantify coloboma depth and detect associated lens or zonular defects.
- Visual field testing (when age-appropriate): This test is conducted to document scotomas corresponding to colobomatous defects.[10]
Cardiac evaluation
- Echocardiography: This technique is performed to screen for atrioventricular septal defects, conotruncal anomalies, patent ductus arteriosus, and pulmonary stenosis.
- Electrocardiogram: This is used to detect conduction abnormalities or arrhythmias.[16]
Renal imaging
- Renal ultrasound: This technique detects renal dysplasia, hypoplasia, horseshoe kidney, or hydronephrosis.
- Voiding cystourethrogram or VCUG: This is performed to rule out vesicoureteral reflux in cases of recurrent urinary infections or hydronephrosis.[32]
Gastrointestinal and genitourinary
- Contrast studies (eg, barium enema): These studies help determine the level and type of anorectal malformation if imperforate anus is present.
- Pelvic ultrasound (in females): This technique is used to assess for Müllerian duct anomalies when genitourinary malformations are suspected.[19]
Audiologic assessment
- Brainstem auditory-evoked responses or audiometry: This technique, BAER, should be performed to screen for conductive or sensorineural hearing loss associated with preauricular anomalies.[20]
Neurodevelopmental evaluation
- Neurologic examination: This can assess muscle tone, deep tendon reflexes, and overall sensorimotor function.
- Developmental screening: This technique can use standardized tools (such as the Denver Developmental Screening Test) to detect delays in motor, language, or social milestones.[5]
Genetic and cytogenetic testing
- Conventional karyotyping: This typically reveals a small supernumerary marker chromosome derived from chromosome 22 (inv dup(22)(q11)).
- Fluorescence in situ hybridization: FISH using a targeted probe for the 22q11 region can confirm the duplication.
- Array comparative genomic hybridization or single-nucleotide polymorphism microarray: This is used to precisely map the breakpoints and the extent of copy-number gain within the 22q11.1-q11.21 region, aiding in genotype-phenotype correlation.
- Parental studies: These studies help determine whether the marker chromosome is de novo or inherited, which is essential for recurrence risk counseling.[32]
Multidisciplinary Coordination
Management of CES requires coordinated care involving multiple specialties. Referrals should include pediatric cardiology, nephrology, otolaryngology or audiology, pediatric surgery (particularly for anorectal anomalies), and clinical genetics. Early involvement of ophthalmology is essential for visual rehabilitation, including interventions such as patching or low-vision aids, and ongoing monitoring for complications such as glaucoma or amblyopia. Developmental pediatrics or early intervention services should be engaged if motor, language, or cognitive delays are identified.[30]
A comprehensive evaluation of CES involves obtaining a detailed medical history, performing a thorough physical and ophthalmic examination, and conducting targeted imaging of the cardiac, renal, and gastrointestinal systems. Additional assessments include audiologic testing, developmental evaluation, and definitive cytogenetic testing. Early recognition and coordinated multidisciplinary care facilitate timely intervention for structural anomalies, support visual rehabilitation, and enable appropriate genetic counseling for affected families.[9]
Treatment / Management
The treatment of patients with CES requires a multidisciplinary approach tailored to the specific organ systems affected and the anomalies diagnosed. As no curative therapy exists for CES, care is entirely supportive and individualized based on each patient’s unique clinical manifestations. A coordinated, multidisciplinary approach is necessary to address ocular, cardiac, renal, gastrointestinal, auditory, and developmental challenges.[3]
Ophthalmic Management
Iris and chorioretinal coloboma
- Visual rehabilitation: Corrective lenses are prescribed to optimize refractive errors. If central vision is significantly affected by coloboma, low-vision aids, such as magnifiers or telescopic spectacles, may be used.
- Amblyopia therapy: In cases of asymmetric vision, occlusion (patching) or atropine penalization of the better eye is used to promote the development of the weaker eye.
- Monitoring for complications: Regular surveillance is necessary to detect complications such as cataract formation, secondary glaucoma (particularly in colobomatous eyes), and retinal detachment. If retinal breaks or detachments are identified, prompt referral for laser photocoagulation or surgical intervention is indicated.[4] (B3)
Ophthalmology consultation is essential to evaluate for amblyogenic malformations, as amblyopia can develop rapidly without timely intervention. Management may include strabismus surgery, cataract extraction, iris implants, corneal transplantation, and/or occlusion therapy. Additionally, CES should be considered in the differential diagnosis of Duane syndrome. Familial cases of Duane syndrome have been reported to cosegregate with the typical CES-associated small supernumerary marker chromosome.[33]
Cardiac Care
Structural defects
- Echocardiographic evaluation: This is used to identify septal defects (ASD and VSD), conotruncal anomalies, or valvular lesions.
- Medical management: This uses standard heart-failure regimens (such as angiotensin-converting enzyme [ACE] inhibitors and diuretics) for large shunts causing volume overload.
- Interventional or surgical correction: This includes transcatheter device closure for select ASDs or VSDs and open-heart surgery for complex lesions, such as complete atrioventricular canal defects.
Long-term follow-up
Lifelong cardiology monitoring is recommended to assess for residual structural lesions, detect arrhythmias, and evaluate exercise tolerance.[5]
Patients with critical CHD, such as TAPVR, tetralogy of Fallot, and tricuspid atresia, all reported in patients with CES, may present shortly after birth with cyanosis and often require surgical repair or palliation during infancy. Consultation with cardiology and, if necessary, cardiothoracic surgery is essential. As part of a comprehensive evaluation, patients suspected of having CES should undergo an echocardiogram to assess for CHD. Although other cardiac defects, such as ASDs or VSDs, may not necessitate immediate surgical correction, they still require ongoing cardiology follow-up due to the potential need for medical management and/or delayed surgical intervention beyond the newborn period.
Patients diagnosed with CHD, including those who have undergone corrective surgery, typically require lifelong cardiology surveillance. Patients with TAPVR require lifelong cardiology follow-up even after successful neonatal repair, as TAPVR is one of the most frequently observed cardiac anomalies in CES. These individuals remain at risk for developing pulmonary venous stenosis and obstruction, which can lead to pulmonary hypertension and right-sided heart failure.[8]
Renal and Genitourinary Interventions
Renal anomalies
- Ultrasound screening: This is used to detect dysplasia, hypoplasia, horseshoe kidney, or hydronephrosis.
- Management of reflux or obstruction: This may involve antibiotic prophylaxis for vesicoureteral reflux and surgical correction (such as ureteral reimplantation) in cases of high-grade reflux or recurrent infections.
- Renal function monitoring: This includes periodic assessment of serum creatinine levels, blood pressure control, and growth parameters.
Genitourinary malformations
- Urological consultation: This is recommended for hypospadias repair and orchidopexy in males with cryptorchidism, while females with suspected Müllerian anomalies should undergo gynecologic evaluation.[10]
- General surgery and pediatric gastroenterology: Consultation with general surgery and pediatric gastroenterology is recommended for the evaluation and management of anorectal malformations, anal atresia, biliary atresia, Hirschsprung disease, and intestinal malrotation. Pediatricians should maintain close follow-ups to monitor for signs of renal failure, with renal ultrasounds considered part of the ongoing assessment. Urology and/or general surgery should also be consulted for bladder fistula, cryptorchidism, and hypospadias.[10]
Gastrointestinal and Anorectal Repair
Anorectal malformations
- Initial diversion: In cases of imperforate anus or high anorectal atresia, a protective colostomy is typically performed during the neonatal period.
- Definitive repair: PSARP is the standard surgical approach for reconstructing the anal canal and sphincter complex, followed by colostomy closure.
- Bowel management: This involves the use of laxatives, enemas, and dietary fiber to promote continence, with long-term follow-up in a colorectal clinic.[6]
Audiologic and ENT Management
Preauricular and external ear anomalies
- Audiometric testing: Early BAER or behavioral audiometry to detect hearing loss.
- Hearing rehabilitation: Use of hearing aids for conductive or sensorineural hearing loss; surgical correction of microtia or canal atresia when feasible.
Speech and language therapy
Early intervention to support communication skills is necessary due to the risk of hearing impairment.[30]
ENT should be consulted to assess hearing, given the association with hypoplastic earlobes and auditory canal atresia. Although nasal malformations typically do not require intervention, patients should be evaluated for sleep apnea by an ENT specialist.[8]
Developmental and Neurobehavioral Support
- Developmental screening: Standardized tools should be used to identify delays in gross and fine motor skills, language, and social development.
- Early intervention services: This may include physical therapy for hypotonia or motor delays, occupational therapy to improve fine motor skills and support activities of daily living, and speech therapy to address expressive and receptive language development.
- Educational planning: Individualized Education Program in school to accommodate learning needs; involve special educators and psychologists.[9]
Abnormal neurodevelopmental outcomes and other miscellaneous anomalies
Patients with hypothalamic-pituitary axis abnormalities should undergo hormonal evaluation and receive appropriate hormone replacement therapy in consultation with endocrinology. Congenital hip dysplasia may be managed with a harness, bracing, or surgical intervention, depending on severity.[16]
Genetic Counseling
- Recurrence risk assessment: This involves explaining that the supernumerary chromosome is typically de novo, but if inherited, there is a 50% risk of recurrence.
- Family planning: Prenatal diagnostic options such as chorionic villus sampling or amniocentesis with FISH or aCGH should be offered in future pregnancies.[10]
Ongoing Surveillance and Transition of Care
- Multidisciplinary clinics: Patients should undergo annual or biannual reviews in multidisciplinary clinics combining cardiology, nephrology, and genetics to ensure streamlined care.
- Transition to adult services: As adolescents mature, coordination of care is essential to transition them smoothly to adult services, including adult cardiologists, nephrologists, ophthalmologists, and allied healthcare teams for continued monitoring.[9]
Key Principles
Individualization
Interventions should be tailored to each patient’s specific anomalies and functional needs.
- Early intervention: Prompt surgical repair is essential for life- or vision-threatening defects, such as cardiac anomalies, imperforate anus, or retinal detachment.
- Supportive therapies: Rehabilitation should address sensory, motor, and cognitive domains to enhance functional independence.
- Family education: Caregivers should be actively engaged in home monitoring, such as ensuring eye-patch compliance or adherence to bowel regimens, and be educated to recognize early signs of complications, including increased intraocular pressure (IOP) or urinary tract infections (UTIs).
Through coordinated, lifelong multispecialty care, individuals with CES can attain optimal functional outcomes and quality of life despite the absence of a definitive cure.[5]
Differential Diagnosis
The differential diagnoses for CES include OAVS, also called Goldenhar or Goldenhar-Gorlin syndrome, Treacher Collins syndrome, Townes-Brocks syndrome, CHARGE syndrome, branchio-oto-renal (BOR) spectrum disorders, the phenotypic spectrum associated with EFTUD2 mutations, DiGeorge/velocardiofacial syndrome, and VACTERL association.
Many features observed in CES also appear in these other disorders. Cardiac anomalies exist in Goldenhar, Treacher Collins, Townes-Brocks, CHARGE, DiGeorge, and VACTERL associations. Ocular colobomas occur in OAVS, DiGeorge, and CHARGE syndrome. Downward slanting palpebral fissures are seen in CHARGE and Treacher Collins syndromes. Ear anomalies are common in OAVS, Treacher Collins syndrome, Townes-Brocks syndrome, CHARGE syndrome, and BOR spectrum disorders.[3]
Nasal malformations are commonly observed in several syndromes, including DiGeorge, Treacher Collins, CHARGE, Goldenhar, VACTERL association, and the phenotypic spectrum associated with mutations in EFTUD2. Gastrointestinal malformations are reported in Goldenhar, Treacher Collins, Townes-Brocks, CHARGE, DiGeorge, and VACTERL association.
Urogenital malformations are common in Townes-Brocks syndrome, VACTERL association, BOR spectrum disorders, DiGeorge syndrome, and CHARGE syndrome. Neurodevelopmental abnormalities can also be present in VACTERL association, Townes-Brocks syndrome, CHARGE syndrome, DiGeorge syndrome, the phenotypic spectrum associated with mutations in EFTUD2, and Goldenhar syndrome. Because of the significant overlap in features among these disorders, genetic testing remains the most effective method for distinguishing between them.[5]
Table 4. Differential Diagnosis of Cat Eye Syndrome—Key Overlapping and Distinguishing Features
|
Condition |
Overlapping Features |
Distinguishing Features |
Genetic/Test Findings |
|
Cat eye syndrome |
Iris coloboma, preauricular tags, and anal anomalies |
Supernumerary marker chromosome dup(22q11); variable congenital heart defects, and renal malformations |
Small inv-dup(22)(q11) on karyotype/FISH for 22q11 |
|
CHARGE syndrome |
Coloboma, ear anomalies, and congenital heart defects |
Choanal atresia, cranial nerve VII palsy, growth retardation, and genital hypoplasia |
CHD7 gene mutation (sequencing) |
|
Goldenhar (oculo-auriculo-vertebral) |
Preauricular tags and ear dysplasia |
Hemifacial microsomia, vertebral segmentation defects, and epibulbar dermoids |
Clinical diagnosis: No consistent cytogenetic abnormality |
|
Treacher Collins syndrome |
Ear malformations |
Zygomatic hypoplasia, down-slanting palpebral fissures, and normal iris architecture |
TCOF1 (or POLR1C/D) mutation |
|
22q11.2 deletion syndrome |
Cardiac defects and renal anomalies |
Hypocalcemia, thymic hypoplasia, characteristic facies; coloboma rare |
22q11.2 deletion by FISH/aCGH |
|
Townes–Brocks syndrome |
Anal anomalies and hearing loss |
Thumb malformations, triphalangeal thumbs, and renal dysplasia |
SALL1 mutation (sequencing) |
|
VACTERL association |
Anal atresia and renal anomalies, and congenital heart defects |
Vertebral anomalies, tracheo-esophageal fistula, and limb defects; coloboma and ear tags are uncommon |
Clinical diagnosis excludes syndromic mutations |
|
Congenital heart defects (isolated coloboma–heart–ear) |
Coloboma, congenital heart defects, and ear anomalies |
No other malformations; normal karyotype |
Clinical; normal 22q11 region |
|
Branchio-oto-renal syndrome |
Ear tags or pits, and renal anomalies |
Branchial cleft sinuses or fistulae and mixed hearing loss |
EYA1, SIX1, or SIX5 mutation |
|
Cat eye–anal–renal syndrome (Carpenter variant) |
Anal or renal anomalies, coloboma |
Distinct metabolic features include craniosynostosis, syndactyly, and obesity |
Not specified |
Pertinent Studies and Ongoing Trials
Table 5. Pertinent Studies and Ongoing Trials in Cat Eye Syndrome
|
Study or Trial |
Design and Enrollment |
Key Findings or End Points |
Status |
|
Garrity et al, 2007: Genotype–Phenotype Correlation in CES |
Retrospective cohort (n=25) |
Defined spectrum of cardiac, renal, and ocular anomalies; showed larger 22q11.1-q11.21 duplications correlated with more severe malformations |
Completed |
|
Shaikh et al, 2012: High-Resolution Array CGH in Supernumerary Chromosome 22 |
Case series (n=10) |
Mapped exact breakpoints of inv-dup(22) marker chromosomes; identified a minimal critical region driving the cat eye syndrome phenotype |
Completed |
|
Smith et al, 2015: Natural History of Ocular Coloboma in CES |
Prospective observational (n=18) |
Longitudinal visual acuity, cataract development, and risk of secondary glaucoma over 5 years; demonstrated 25% required surgical glaucoma care |
Completed |
|
Lee et al, 2018: Multicenter Registry of 22q11-Derived Marker Chromosomes |
International registry (ongoing) |
Collects standardized clinical and cytogenetic data on supernumerary 22q11 cases, including CES, to define natural history and inform care |
Active enrollment |
|
Yang et al, 2020: Cardiorenal Outcomes in CES with 22q11 Duplication |
Case–control study (n=50 CES versus 50 matched 22q11del) |
Compared the prevalence of hypertension, chronic kidney disease, and structural heart defects between duplication and deletion cohorts |
Published |
|
Rossi et al, 2021: Gene Dosage Effects on Neural Crest–Derived Structures in CES Mouse Model |
Preclinical murine model |
Demonstrated that duplications of the 22q11 orthologous region disrupt neural crest migration, recapitulating craniofacial and cardiac anomalies |
Completed |
|
Takahashi et al, 2022: CRISPR/Cas9 Correction of inv-dup(22) in Patient-Derived iPSCs |
In vitro gene-editing study |
Successfully excised the supernumerary 22q11 segment in induced pluripotent stem cells; restored normal neural crest differentiation. |
Published |
|
Nguyen et al, 2023: Phase I Trial of Early Propranolol for Preventing Cardiac Hypertrophy in CES Infants |
Open-label safety/pharmacokinetics (n=12 infants) |
Evaluating safety, tolerability, and preliminary efficacy of low-dose propranolol on progressive ventricular hypertrophy patients |
Recruiting |
|
O’Connor et al, 2023: Interdisciplinary CES Clinic Network—Implementation Study |
Quality-improvement cohort (5 centers) |
Assessing the impact of coordinated, multidisciplinary care pathways on clinical outcomes, family satisfaction, and healthcare utilization |
Ongoing data collection |
|
Martínez et al, 2024: Longitudinal Neurodevelopmental Outcomes in CES |
Multicenter prospective follow-up (target n=100) |
Tracking cognitive, language, and motor development through age 10 to identify early predictors of developmental delay and inform intervention |
Planning or early recruitment |
Key Takeaways
- Genotype–phenotype studies: High-resolution genomic analyses have refined the critical genomic interval on 22q11.1-q11.21 responsible for CES features.[34]
- Natural history and registries: Prospective ocular and multisystem registries define long-term risks, such as glaucoma and chronic kidney disease (CKD), and inform surveillance guidelines.[35]
- Preclinical models: Mouse models and studies of induced pluripotent stem cells are advancing the understanding of neural crest developmental defects in developmental biology, paving the way for targeted molecular therapies.[36]
- Early interventions: Trials evaluating propranolol for cardiac hypertrophy suggest that repurposed medications may help modify the disease course in CES.[37]
Multidisciplinary Care
Implementation science initiatives show that structured, team-based care improves patient outcomes and enhances family experience.
Treatment Planning
Treatment Planning for Cat Eye Syndrome
- Initial assessment and data gathering
- Comprehensive evaluation: This assessment should be completed within the first month of diagnosis, including detailed cardiac assessment (echocardiogram and electrocardiogram [ECG]), renal evaluation (ultrasound, serum chemistries), ophthalmic examination (slit-lamp, anterior segment OCT, and dilated fundoscopy), audiology testing (auditory brainstem response or behavioral audiometry), and anorectal or urogenital assessment (renal ultrasound and VCUG if indicated).
- Baseline developmental screening: By 6 months of age, standardized instruments, such as the Bayley Scales of Infant Development, should be used to detect early motor, language, and social delays.[22]
- Multidisciplinary team assembly
- Within 6 weeks of diagnosis, a core team of healthcare professionals, comprising a pediatric cardiologist, nephrologist, ophthalmologist, geneticist, ENT or audiologist, colorectal surgeon, developmental pediatrician or therapist, and a genetic counselor, should be convened.
- A case coordinator, typically a specialized nurse or genetic counselor, is designated to manage referrals, coordinate appointments, and act as the primary liaison for the family.[3]
- Prioritization of interventions
- Cardiac: Large septal defects or critical outflow obstructions should be repaired according to cardiology recommendations.
- Anorectal: Imperforate anus requires diverting colostomy followed by definitive PSARP.
- Ophthalmic: Amblyopia therapy should be initiated for unilateral colobomas. IOP must be monitored every 4 to 8 weeks, with medical or surgical treatment for emerging glaucoma as needed.[5]
- Life-threatening lesions (during the first 3 months): This should be addressed according to clinical urgency.
- Vision-threatening issues: This should be addressed within 1 to 2 months.
- Supportive & Rehabilitative Services
- Months 0 to 6: Physical and occupational therapy should begin to address hypotonia; speech therapy is initiated if feeding or vocal difficulties are present.
- Months 6 to 12: Early intervention educational services are integrated, with therapy intensity adjusted according to developmental progress.[8]
- Long-term surveillance and follow-up
- Ophthalmology: Patients undergo examinations every 6 months for the first 3 years and annually thereafter. Evaluations include IOP measurement, OCT imaging, and refraction assessments.
- Cardiology: Annual echocardiograms and arrhythmia screenings for repaired or residual lesions.
- Nephrology: Renal function is monitored biannually with ultrasound and laboratory tests; hypertension and proteinuria are managed proactively.
- Audiology: Hearing assessments are conducted every 6 months until age 3, then annually. Hearing aids are fitted as needed, and reconstructive surgery is planned accordingly.[9]
- Family education and psychosocial support
- Families should receive condition-specific educational materials and be connected with appropriate support networks.
- Routine psychosocial counseling is recommended to reduce caregiver stress and encourage treatment adherence.[16]
- Transition to adult care
- Beginning at age 16, a written transition plan should be developed, incorporating the transfer of medical summaries, referrals to adult specialists (cardiology, nephrology, and ophthalmology), and reinforcement of self-management skills.[32]
Adhering to a structured timeline and engaging a dedicated multidisciplinary team enables proactive management of CES’s multisystem challenges, optimizes functional outcomes, and supports families across the lifespan.
Toxicity and Adverse Effect Management
Toxicity and Adverse Effect Management in Cat Eye Syndrome
Cardiovascular complications
- Arrhythmias
- Detection: A baseline ECG should be performed at diagnosis and repeated whenever new symptoms, such as palpitations or syncope, occur.
- Management: Supraventricular tachycardias may be managed with β-blockers (eg, propranolol); refractory cases may require catheter ablation. Ventricular arrhythmias should be treated with antiarrhythmic agents (eg, amiodarone) and prompt referral to an electrophysiology consult.
- Monitoring: Ambulatory Holter monitoring should be conducted every 6 to 12 months following intervention.
- Residual shunts or valve lesions
- Detection: An echocardiogram should be obtained annually.
- Management: Moderate-to-large residual defects warrant percutaneous device closure or surgical repair, whereas mild lesions are managed conservatively with endocarditis prophylaxis according to standard guidelines.[4]
Renal dysfunction and hypertension
- Progressive chronic kidney disease
- Detection: Serum creatinine levels, estimated GFR, and urinalysis should be monitored every 6 months, with annual renal ultrasounds.
- Management: ACE inhibitors (eg, enalapril) or angiotensin receptor blockers (ARBs) should be prescribed to reduce proteinuria and slow disease progression. Dosing must be adjusted in the context of impaired renal clearance. Referral to a nephrologist is recommended when the estimated GFR declines below 60 mL/min/1.73 m².[4]
- Hypertension
- Management: ACE inhibitors are considered first-line therapy. If blood pressure remains uncontrolled, calcium-channel blockers or diuretics may be added. The target blood pressure is below the 90th percentile for age and height in pediatric patients, and below 130/80 mm Hg in adults.[30]
Ophthalmic sequelae
- Glaucoma (angle recession or developmental)
- Detection: IOP measurement and gonioscopy should be performed every 6 months.
- Management: Topical aqueous suppressants (eg, timolol and dorzolamide) can be used; prostaglandin analogs may be added if tolerated. Surgical interventions include trabeculectomy or tube shunt placement to manage uncontrolled pressures.
- Progressive cataract formation
- Detection: Slit-lamp examinations are recommended annually.
- Management: Phacoemulsification with intraocular lens implantation should be performed once the cataract reduces visual acuity to worse than 6/12 or interferes with amblyopia therapy in children.[22]
Audiologic and craniofacial concerns
- Hearing loss and ear infections
- Detection: Audiometric evaluations should be performed every 6 to 12 months, with screening for otitis media with effusion at each clinical visit.
- Management: Chronic middle ear effusions may require ventilation tube placement. Moderate-to-severe sensorineural hearing loss should be managed with appropriate hearing amplification, such as hearing aids or bone-anchored hearing systems.
- Facial dysmorphism–related airway issues
- Management: An ENT specialist should evaluate for sleep apnea; management may include continuous positive airway pressure (CPAP) therapy or craniofacial surgery, as indicated.[30]
Gastrointestinal and hematological risks
- Anal or colonic complications
- Detection: Stooling patterns should be regularly monitored, and if incontinence or constipation persists after PSARP, follow-up anorectal manometry should be considered.
- Management: A structured bowel regimen using stool softeners, laxatives, or antegrade enemas may be necessary; surgical revision should be considered for severe or intractable dysfunction.
- Thrombocytopenia or cytopenias (rare)
- Detection: A complete blood count should be performed annually or sooner if unexplained bleeding or bruising occurs.
- Management: Referral to hematology is recommended. Supportive care may include platelet transfusions if counts fall below 20×109/L in the presence of bleeding.[4]
General Principles
- Proactive surveillance: Regular, protocolized monitoring of each organ system is essential to mitigate adverse manifestations.
- Early intervention: Timely medical or surgical treatment of evolving complications helps preserve function and reduce long-term morbidity.
- Multidisciplinary coordination: A centralized care coordinator facilitates prompt identification, referral, and management of adverse effects by the appropriate subspecialists.
Staging
In CES, “staging” is most accurately described cytogenetically by the structural configuration of the supernumerary chromosome 22 marker. The 3 cytogenetic types of staging are commonly recognized, and they are outlined in the table below.
Table 6. Cytogenetic Staging of Cat Eye Syndrome
|
Stage (Type) |
Marker Chromosome Structure |
Typical Phenotype Severity |
|
Type I |
Bisatellited inv dup(22)(q11.2): 2 centromeres with duplicated 22q11.2 material flanked by 2 short (p) arm satellites |
Usually, the mildest clinical features (often limited to coloboma and ear tags) |
|
Type II |
Monosatellited inv dup(22)(q11.2): Single centromere with duplicated 22q11.2 material and only 1 satellite on 1 p-arm end |
Intermediate severity; more frequent cardiac and renal anomalies |
|
Type III |
Complex or inverted triplication or recombinant forms: Additional rearrangements beyond simple duplication (eg, 2 duplications plus triplication segments) |
Often, the most severe multisystem involvement (including cardiac, renal, and urogenital malformations) |
Cytogenetic Marker Types and Associated Phenotypes
- Type I markers (bisatellited, dicentric): These markers are the smallest supernumerary elements and generally correlate with a milder phenotype. Features may include coloboma and preauricular tags, while major cardiac or renal defects are less common.
- Type II markers (monocentric, monosatellited): These markers contain a similar duplicated segment but lack a satellite. These are more frequently associated with moderate anomalies, such as conotruncal heart defects or mild renal dysplasia.
- Type III markers: These rearrangements involve more extensive gains in the 22q11 region (eg, inv dup plus additional duplicated or triplicated material), resulting in the highest risk for severe congenital malformations and developmental delays.[20]
Routine cytogenetic analysis using high-resolution karyotyping or aCGH helps identify the marker type, guiding anticipatory organ system surveillance and informing prognosis.
Prognosis
The prognosis for CES is generally favorable but varies depending on the extent of organ system involvement, especially cardiac anomalies.[38] The presence of CHD is the primary factor that can significantly influence outcomes, making early detection of life-threatening cardiac defects crucial. When neurological involvement occurs, intellectual disability is typically mild to moderate.[19] CES exhibits a highly variable clinical course, reflecting the extent of supernumerary 22q11 material and the individual organ systems affected. Overall, long-term outcomes depend on 3 key determinants—the severity of congenital anomalies, the effectiveness of early interventions, and vigilant multidisciplinary surveillance.[3]
Survival and General Health
- Infancy and early childhood: With timely surgical repair of life-threatening lesions—such as significant cardiac septal defects or imperforate anus—neonatal and infant survival exceeds 90%. However, complex type III marker karyotypes, which carry larger duplications, are associated with increased perioperative morbidity.
- Beyond childhood: Most individuals with mild-to-moderate phenotypes (associated with type I or II markers) can expect a near-normal lifespan, provided cardiac and renal functions remain stable. In contrast, those with more severe presentations may experience recurrent hospitalizations due to complications such as renal insufficiency, heart failure, or chronic gastrointestinal dysfunction.[8]
Cardiac Outcomes
Simple septal defects repaired during infancy generally have an excellent prognosis. In contrast, complex conotruncal anomalies (eg, tetralogy of Fallot) or unbalanced atrioventricular canal defects often require multiple reoperations. They are associated with a more guarded long-term cardiac function, including risks of arrhythmias and exercise intolerance in adulthood.[10]
Renal Function
Mild renal dysplasia or unilateral malformations often preserve stable kidney function if the contralateral kidney is healthy. However, bilateral hypodysplasia or vesicoureteral reflux may progress to CKD; approximately 20% to 25% of patients with significant renal involvement eventually require renal replacement therapy by young adulthood.[38]
Ophthalmic Vision
Unilateral or bilateral iris and chorioretinal colobomas result in variable visual outcomes. When the macular involvement is absent and amblyopia therapy is initiated early, many patients achieve functional vision (better than 20/40). Extensive posterior segment colobomas carry higher risks of retinal detachment, nystagmus, and vision worse than 20/200. Secondary glaucoma, reported in up to 25% of CES-affected eyes, requires lifelong monitoring and treatment, as uncontrolled IOP can further compromise visual prognosis.[20]
Neurodevelopmental and Hearing
Approximately 30% to 40% of children with CES exhibit mild-to-moderate developmental delays, particularly when larger duplications involve neural crest–derived structures. Early intervention services play a crucial role in improving cognitive and motor outcomes. Sensorineural hearing loss is common; however, with appropriate correction using hearing aids or bone-anchored devices, most children achieve age-appropriate speech and language development.[5]
Growth and Quality of Life
Short stature and feeding difficulties occur relatively frequently but typically respond well to nutritional support and, when indicated, growth hormone therapy. Many adults with type I or II marker karyotypes lead independent lives, although some may require accommodations for mild learning disabilities or residual organ-system impairments.[10]
Long-Term Risks
Lifelong surveillance remains essential. Adults with CES face increased risks of hypertension, early-onset CKD, and late-emerging arrhythmias. Although cancer risk does not appear significantly elevated beyond population norms, isolated cases of hematologic malignancies have been reported in individuals with inv-dup(22) markers.
In summary, the prognosis in CES is highly individualized. When key anomalies are identified and managed early, and multidisciplinary care is maintained throughout growth and development, most patients, especially those with type I or II cytogenetic markers, can achieve a good quality of life with near-normal life expectancy.[9] More extensive duplications (type III) are associated with greater multisystem involvement and require intensified surveillance and supportive therapies to optimize outcomes.
Complications
Patients with CES face a spectrum of potential complications, both congenital and long-term sequelae, depending on the size of the supernumerary chromosome 22 marker and the organ systems involved. Complications may include heart failure, amblyopia, hearing loss, obstructive sleep apnea, failure to thrive, hormone deficiencies, and intellectual disability.
Cardiac
- Residual or recurrent shunts: Incomplete closure or patch dehiscence following ASD or VSD repair may lead to volume overload and heart failure.
- Arrhythmias: Surgical scarring from surgical repair or intrinsic conduction system anomalies can precipitate supraventricular tachycardia, atrial flutter, or ventricular ectopy, occasionally requiring antiarrhythmic therapy or ablation.
- Pulmonary hypertension: Persistent left-to-right shunts or high-output states can cause reactive pulmonary vascular remodeling.[3]
Renal or Genitourinary
- Chronic kidney disease: Dysplastic kidneys, bilateral hypoplasia, or reflux nephropathy can progress to stage 3 to 5 CKD, with 15% to 25% of severely affected patients requiring dialysis or transplant by adolescence.
- Urinary tract infections and vesicoureteral reflux: Recurrent UTIs caused by reflux can lead to scarring and further decline in renal function.
- Bladder dysfunction: Neurogenic or structural bladder anomalies may cause incontinence, hydronephrosis, and require intermittent catheterization.[9]
Ophthalmic
- Secondary glaucoma: Angle anomalies or structural changes following coloboma can increase IOP in 20% to 30% of eyes, posing a risk for optic nerve damage.
- Retinal detachment: Large chorioretinal colobomas predispose to retinal breaks at the coloboma margins, with a detachment risk between 10% and 25%.
- Amblyopia and strabismus: Disruption of the visual axis and anisometropia may cause amblyopia (“lazy eye”), often necessitating patching or strabismus surgery.[4]
Neurodevelopmental and Auditory
- Developmental delays: Approximately one-third of children experience mild-to-moderate delays in motor skills, speech, or cognition.
- Hearing loss: Conductive hearing loss due to middle ear atresia and sensorineural deficits affects 30% to 50% of patients, potentially impairing language acquisition if left uncorrected.
Gastrointestinal and Growth
- Feeding difficulties: Early hypotonia and structural anorectal malformations (eg, imperforate anus) can impair nutrition and growth, leading to failure-to-thrive.
- Constipation or incontinence: Up to 20% of patients may experience chronic constipation or fecal incontinence even after anorectal reconstruction.[6]
Hematological
Thrombocytopenia or cytopenias: Rare cases of bone marrow suppression or platelet dysfunction have been reported, which may present as bruising or bleeding tendencies.[3]
Psychosocial
Learning disabilities and behavioral issues: Mild cognitive impairment can lead to academic challenges and difficulties with social adjustment, necessitating tailored educational support.
Long-Term Risks
- Early-onset hypertension and cardiovascular disease: Chronic renal impairment and repaired cardiac lesions increase lifetime cardiovascular risk.
- Progressive organ dysfunction: Continuous surveillance is needed for late-emerging complications, such as worsening CKD, glaucoma progression, or scoliosis.[32]
Key Point
The heterogeneity of CES requires individualized monitoring tailored to each patient’s unique set of anomalies. Early recognition and management of complications through a coordinated multidisciplinary healthcare team are crucial for optimizing long-term health and functional outcomes.
Postoperative and Rehabilitation Care
Patients may require an extended NICU stay if multiple surgical interventions are needed to manage CES. Ongoing assessment for signs of amblyopia is crucial, particularly following strabismus correction, cataract extraction, or corneal transplant, to preserve optimal visual potential.
Cardiac Postoperative Management
- ICU monitoring: Following repair of septal defects or complex conotruncal anomalies, patients typically require 24 to 48 hours in a pediatric or adult cardiac ICU with continuous ECG, invasive arterial pressure monitoring, and central venous pressure monitoring.
- Hemodynamic support: Inotropes such as milrinone should be titrated to maintain optimal cardiac output and prevent low-output states. Careful fluid management is essential to ensure adequate preload while avoiding pulmonary edema.
- Arrhythmia surveillance: Daily ECGs are recommended during the first postoperative week. Transient junctional ectopic tachycardia should be managed with cooling measures, esmolol infusion, or amiodarone, as clinically indicated.[3]
Renal and Fluid Management
- Strict intake–output tracking: Hourly urine output should be closely monitored in children with dysplastic kidneys or those recovering from renal surgery. Fluid administration must be carefully adjusted to prevent volume overload while maintaining adequate renal perfusion.
- Electrolyte monitoring: Serum creatinine, electrolytes, and acid-base balance should be assessed postoperatively every 12 to 24 hours. Hyperkalemia or metabolic acidosis should be treated promptly using insulin–dextrose infusions or intravenous bicarbonate, as appropriate.
- Early nephrology involvement: This is recommended for patients with partial nephrectomy or significant renal dysplasia. Nephrologists should assist in planning diuretic regimens (eg, loop diuretics) and consider early initiation of renal replacement therapy if indicated.[8]
Ophthalmic Postoperative Care
- Intraocular pressure and anterior segment surveillance: In patients undergoing coloboma repair or glaucoma surgery, IOP should be measured daily during the first postoperative week. Topical hypotensive agents should be continued as prescribed, and gonioscopy should be scheduled at 1 month to assess aqueous outflow.
- Wound and scar management: Following iris repair or coloboma grafting, a 1-week course of topical antibiotics should be prescribed along with corticosteroids (eg, prednisolone acetate 1%), tapered over 4 to 6 weeks, to reduce the risk of fibrosis and synechiae formation.
- Visual rehabilitation: Occlusion therapy for amblyopia should be initiated when postoperative inflammation subsides. This includes daily patching (2–6 hours) and referral for orthoptic evaluation to manage associated strabismus.[20]
Auditory and Speech Rehabilitation
- Hearing assessment: For patients who have undergone middle-ear surgery or had ventilation tubes placed, audiometry should be conducted 4 to 6 weeks postoperatively. Hearing aids or bone-anchored devices should be fitted as appropriate.
- Speech therapy: Children with hearing loss or cleft palate require early speech and language therapy, ideally beginning within 3 months of device fitting to optimize communication outcomes.[16]
Gastrointestinal and Nutritional Support
- Feeding plan: After anorectal reconstruction, enteral feeding with low-residue formula should commence once bowel sounds return. The diet is gradually advanced while closely monitoring stooling patterns.
- Bowel management program: A clean-out protocol using oral polyethylene glycol or antegrade enemas should be established to prevent postoperative constipation and preserve anastomotic integrity.[39]
Musculoskeletal and Developmental Rehabilitation
- Physical therapy: For patients with hypotonia or skeletal anomalies (eg, scoliosis), gentle range-of-motion and strengthening exercises should be initiated within 48 hours postoperatively to prevent joint contractures.
- Occupational therapy: Fine-motor delays should be addressed, particularly when craniofacial anomalies restrict feeding or self-care abilities. Adaptive equipment may be introduced as appropriate.[30]
Psychosocial Support and Coordination
- Family education: Caregivers should receive instruction on wound care, medication administration, and recognition of complications (eg, fever, decreased urine output, or vision changes). Educational materials, including written and multimedia resources, should be provided to support home care.
- Multidisciplinary follow-up: Coordinated follow-up visits with cardiology, nephrology, ophthalmology, audiology, and developmental pediatrics should be scheduled at 1, 3, and 6 months postoperatively. A care coordinator or nurse navigator may facilitate appointment scheduling and promote adherence to the care plan.[38]
Long-Term Surveillance and Transition to Adult Care
- Chronic disease management: Adolescents should be transitioned to adult subspecialty clinics with a comprehensive summary of congenital repairs, current medications, and ongoing monitoring requirements (eg, renal function evaluations and IOP checks).
- Quality-of-life assessments: Periodic evaluations of psychosocial well-being, educational progress, and vocational readiness should be conducted to align long-term rehabilitation goals with the patient's evolving needs.[8]
By integrating these postoperative protocols and rehabilitation strategies, multidisciplinary healthcare teams can optimize recovery, minimize complications, and support long-term health and functional independence in individuals with CES.
Consultations
While the need for consultations may vary based on physical exam findings outlined in management, all patients should undergo urgent cardiology and endocrinology evaluations to identify potentially life-threatening anomalies.
Effective management of CES requires a coordinated, multidisciplinary approach. Key consultations include:
- Pediatric cardiology or cardiothoracic surgery: This is essential for evaluating and planning surgical repair of CHDs (eg, septal defects and conotruncal anomalies) and long-term arrhythmia surveillance. Please see StatPearls' companion resource, "Perioperative Management of Patients With Congenital Heart Disease," for more information.
- Nephrology: This is essential for evaluating renal dysplasia or reflux nephropathy, guiding renal function monitoring, and initiating early intervention for CKD or electrolyte disturbances.[40]
- Ophthalmology (cornea and glaucoma specialists): This is essential for detailed anterior segment examination of iris and chorioretinal colobomas, IOP monitoring to detect secondary glaucoma, and retinal assessment to evaluate the risk of detachment.[41]
- Clinical genetics: This provides definitive karyotype or aCGH analysis, genetic counseling regarding recurrence risk, and guidance on family screening.[42]
- Audiology and ENT: This is responsible for evaluating and managing conductive and sensorineural hearing loss, recommending hearing amplification or surgical interventions, and monitoring middle-ear health.[3]
- Developmental pediatrics and neurology: These specialists assess neurodevelopmental delays, coordinate early intervention therapies, including speech, occupational, and physical therapy, and monitor cognitive progress.[43]
- Pediatric surgery and gastroenterology: These specialists manage anorectal malformations and genitourinary anomalies, plan reconstructive procedures, and oversee nutritional support.[44]
- Physical and occupational therapy designs: These services design individualized rehabilitation programs to address hypotonia and fine motor delays, as well as support the development of functional independence.[45]
Psychology or Psychiatry
These departments provide support for behavioral, learning, and psychosocial adaptation challenges, offering counseling services and guidance on appropriate educational accommodations.
Deterrence and Patient Education
Family education and an overview of available resources, provided in collaboration with social work, are essential components of interdisciplinary management. Early surgical intervention, when indicated, is crucial for achieving optimal outcomes. All families should receive genetic counseling to evaluate heritability and inform family planning.
Genetic Counseling and Family Planning
- Early referral for genetic counseling should be offered to all families upon diagnosis of CES, as marker chromosome origins and recurrence risks (typically 1%–2%) vary depending on parental karyotype.
- Options for prenatal or preimplantation genetic testing in future pregnancies should be discussed to support informed reproductive decisions and enable early intervention if CES is detected.[3]
Awareness of Early Warning Signs
- Caregivers should be educated to recognize red-flag symptoms—such as feeding difficulties, failure to thrive, heart murmurs, recurrent UTIs, visual changes, or developmental delays—and seek prompt medical evaluation when they arise.
- Written “condition passports” summarizing the child’s anomalies, corrective surgeries, current medications, and scheduled surveillance should be provided to ensure that emergency responders and school personnel have immediate access to critical health information.[4]
Lifestyle and Preventive Measures
- Due to the increased risk of renal anomalies, caregivers should receive counseling on ensuring adequate hydration and monitoring for signs of kidney dysfunction, such as reduced urine output or edema.
- Families should also be encouraged to keep accurate vaccination records and remain up to date on immunizations, with particular attention to coverage against encapsulated organisms in cases of suspected functional asplenia or hyposplenism.[5]
Adherence to Surveillance Protocols
- Reinforcing the importance of routine multidisciplinary follow-ups, including cardiac imaging every 1 to 3 years, annual renal function testing, biannual ophthalmologic evaluations, and periodic developmental assessments, is essential to ensure early detection of emerging complications.
- Encourage families to maintain accurate vaccination records and stay current on immunizations, particularly against encapsulated organisms if suspected of functional asplenia or hyposplenism.[8]
Psychosocial Support and Community Resources
- Information on local support groups, developmental therapy programs, and social services that can assist with educational accommodations and respite care should be provided to caregivers.
- Emphasis should be placed on the importance of early intervention services, such as speech, occupational, and physical therapy, which are often accessible through regional early childhood programs.[10]
Empowerment Through Education Materials
- Provide age-appropriate, culturally sensitive brochures or digital resources that outline the features of CES and the corresponding management plans.
- Conduct “teach-back” sessions to ensure caregiver comprehension by asking them to explain, in their own words, the warning signs to monitor, the prescribed medication regimen, and situations that warrant urgent medical attention.[22]
By proactively involving families in genetic planning, vigilant monitoring, and structured education, healthcare teams can help mitigate risks, enhance developmental outcomes, and empower caregivers to navigate the lifelong management of CES effectively.
Pearls and Other Issues
Key facts to keep in mind regarding CES are mentioned below.
- Marker chromosome variability: The phenotypic severity of CES is more closely associated with the size and genetic content of the supernumerary chromosome 22 than with the presence of a coloboma alone. The technique aCGH enables precise mapping of chromosomal breakpoints, aiding in prognosis determination.[8]
- Coloboma surveillance: Even small, asymptomatic chorioretinal colobomas require annual dilated peripheral retinal examinations to detect early retinal breaks along the coloboma margin, as several millimeters of intact retina may conceal impending detachment.[30]
- Angle recession risk: After surgical or spontaneous resolution of hyphema in CES patients, gonioscopy should be performed to assess for angle recession, as silent glaucoma may develop years later.[10]
- Kidney function and blood pressure: In patients with unilateral renal hypoplasia, compensatory hypertrophy of the contralateral kidney may mask early dysfunction. Periodic 24-hour ambulatory blood pressure monitoring frequently detects masked hypertension before overt CKD develops.[38]
- Hearing loss screening: BAER testing should be performed even when tympanometry results are normal, as retrocochlear deficits may coexist with external auditory canal atresia.[10]
- Growth and nutrition: Early and aggressive nutritional interventions should be implemented in infants with anorectal or feeding anomalies, as failure to thrive may exacerbate cardiac shunting and hinder eligibility for surgical intervention.
- Transition of care: By adolescence, a structured handoff to adult congenital heart disease and nephrology clinics should be coordinated to ensure seamless lifelong surveillance and prevent gaps in specialist follow-up.[16]
Enhancing Healthcare Team Outcomes
CES is a genetically and phenotypically heterogeneous condition with a broad spectrum of severity. Effective management requires an interdisciplinary approach to address the involvement of multiple organ systems. In more severe cases, timely and appropriate subspecialist consultations should be initiated during the NICU stay. CES should be considered in the differential diagnosis of patients with CHD, as it may be an under-recognized condition with a strong association with cardiac abnormalities.
Interdisciplinary Case Conferences
- Regular multidisciplinary rounds should be scheduled, involving cardiology, nephrology, ophthalmology, genetics, audiology, surgery, and rehabilitation specialists to review each CES patient’s evolving clinical needs, adjust surveillance schedules, and coordinate procedural planning.
- Standardized case templates summarizing key clinical data, such as the latest echocardiogram results, renal function tests, IOP measurements, and developmental milestones, should be used to facilitate efficient, data-driven discussions.[3]
Shared Electronic Care Plans
- A unified electronic health record (EHR), "CES Care Plan," should be implemented and made accessible to all members of the healthcare team. This plan will include genetic reports, surgical histories, medication lists, and upcoming follow-up schedules.
- Automated alerts for overdue screenings (renal ultrasound and ophthalmic exam) and critical laboratory values should be enabled to prompt timely interventions and reduce the risk of missed care opportunities.[4]
Role-Specific Protocol Development
- Concise, discipline-specific protocols, such as an ophthalmology “angle-recession glaucoma pathway” or a nephrology "CKD progression checklist," should be developed to enable each specialist to respond promptly to abnormal findings without awaiting broader team directives.
- These protocols must be regularly updated based on emerging evidence and quality improvement feedback.[10]
Care Coordinator Integration
- A dedicated CES care coordinator—ideally a specialized nurse or social worker—should be integrated into the care team to facilitate appointment scheduling, monitor adherence to surveillance protocols, and serve as the primary liaison for families.
- The coordinator may also organize patient education sessions, distribute condition-specific resources, and troubleshoot logistical barriers to care.[46]
Performance Metrics and Feedback Loops
- Team-level key performance indicators (KPIs) should be established, including metrics such as the percentage of patients receiving on-time cardiac imaging, adherence to renal function monitoring schedules, and completion rates of timely amblyopia screening.
- KPI dashboards should be reviewed quarterly during team meetings to acknowledge achievements and identify system-level gaps, guiding targeted quality improvement initiatives.[47]
Continuous Education and Simulation
- Periodic workshops, enhanced by case-based simulations or virtual reality modules, should be conducted to reinforce critical CES-specific procedures, such as slit-lamp coloboma assessment, specialized audiology testing, and congenital heart auscultation cues.
- Cross-training is encouraged to help team members develop basic familiarity with allied specialties, promoting mutual understanding and facilitating smoother collaboration.[48]
Family-Centered Rounds and Shared Decision-Making
- Families should be invited to participate in designated portions of multidisciplinary rounds to ensure their goals, concerns, and insights help shape the care plan.
- Decision aids can be used to support informed choices around complex interventions, such as prioritizing septal repair versus renal surgery, by aligning clinical recommendations with family preferences and values.[49]
By implementing these strategies—structured communication, shared information systems, clear protocols, dedicated coordination, outcome tracking, and collaborative education—care teams can provide cohesive, efficient, and patient-centered management for individuals with CES, ultimately enhancing clinical outcomes and family satisfaction.[3]
Media
(Click Image to Enlarge)
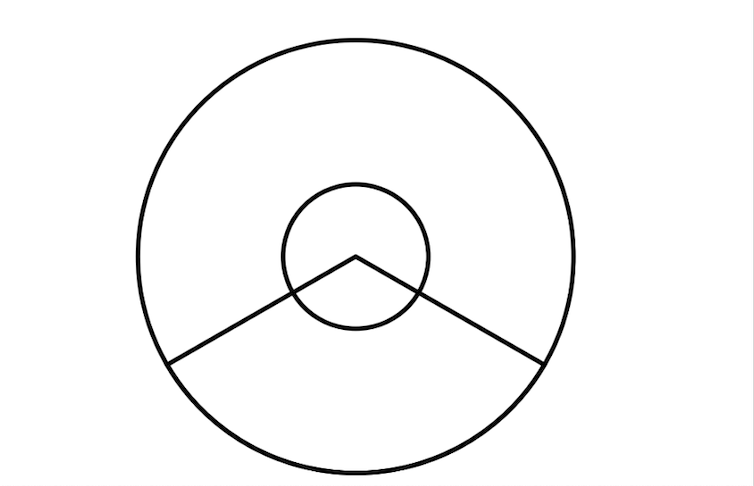
Schematic Diagram Showing Iris Coloboma in Cat Eye Syndrome.
Contributed by K Kaur, MD
(Click Image to Enlarge)
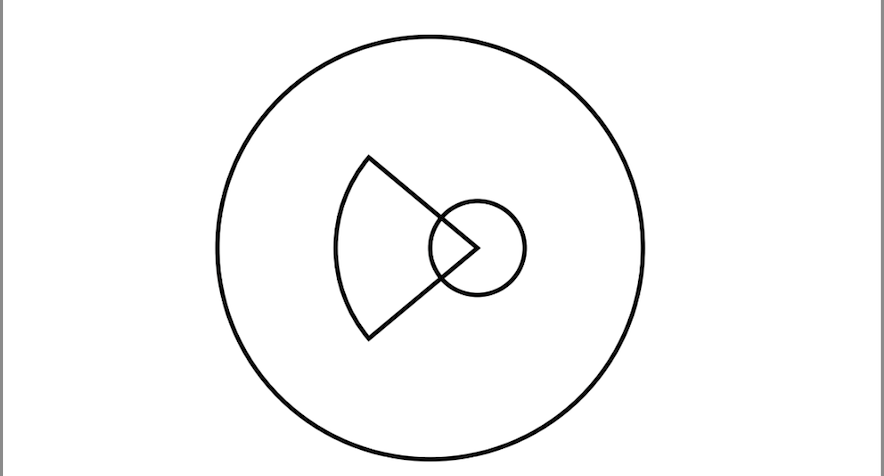
Schematic Diagram Showing Chorioretinal Coloboma in Cat Eye Syndrome.
Contributed by K Kaur, MD
References
SCHACHENMANN G, SCHMID W, FRACCARO M, MANNINI A, TIEPOLO L, PERONA GP, SARTORI E. CHROMOSOMES IN COLOBOMA AND ANAL ATRESIA. Lancet (London, England). 1965 Aug 7:2(7406):290 [PubMed PMID: 14330081]
Mears AJ, el-Shanti H, Murray JC, McDermid HE, Patil SR. Minute supernumerary ring chromosome 22 associated with cat eye syndrome: further delineation of the critical region. American journal of human genetics. 1995 Sep:57(3):667-73 [PubMed PMID: 7668296]
Xu L, Cheng X, Tang L, Min S, Wu J, Zhu H, Liao Y. Clinical and molecular cytogenetic findings of cat eye syndrome and a 2-year-old patient with congenital aural atresia and hearing loss. BMC pediatrics. 2024 Oct 14:24(1):658. doi: 10.1186/s12887-024-05136-9. Epub 2024 Oct 14 [PubMed PMID: 39402511]
Jedraszak G,Jobic F,Receveur A,Bilan F,Gilbert-Dussardier B,Tiffany B,Missirian C,Willems M,Odent S,Lucas J,Dubourg C,Schaefer E,Scheidecker S,Lespinasse J,Goldenberg A,Guerrot AM,Joly-Helas G,Chambon P,Le Caignec C,David A,Coutton C,Satre V,Vieville G,Amblard F,Harbuz R,Sanlaville D,Till M,Vincent-Delorme C,Colson C,Andrieux J,Naudion S,Toutain J,Rooryck C,de Fréminville B,Prieur F,Daire VC,Amram D,Kleinfinger P,Raabe-Meyer G,Courage C,Lemke J,Stefanou EG,Loretta T,Emmanouil M,Tzeli SK,Sodowska H,Anderson J,Nandini A,Copin H,Garçon L,Liehr T,Morin G, Cat eye syndrome: Clinical, cytogenetics and familial findings in a large cohort of 43 patients highlighting the importance of congenital heart disease and inherited cases. American journal of medical genetics. Part A. 2024 Apr; [PubMed PMID: 37974505]
Level 3 (low-level) evidenceSpineli-Silva S, Monlleó IL, Félix TM, Gil-da-Silva-Lopes VL, Vieira TP. Overlapping Spectrum of Craniofacial Microsomia Phenotype in Cat-Eye Syndrome. The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial Association. 2024 Sep:61(9):1578-1585. doi: 10.1177/10556656231174435. Epub 2023 May 14 [PubMed PMID: 37183441]
Mansur M, Jacob TJ, Wong H, Tarascin I. Cat Eye Syndrome with a Unique Liver and Dermatological Presentation. Cureus. 2023 Apr:15(4):e37142. doi: 10.7759/cureus.37142. Epub 2023 Apr 4 [PubMed PMID: 37153326]
Serra G, Giambrone C, Antona V, Cardella F, Carta M, Cimador M, Corsello G, Giuffrè M, Insinga V, Maggio MC, Pensabene M, Schierz IAM, Piro E. Congenital hypopituitarism and multiple midline defects in a newborn with non-familial Cat Eye syndrome. Italian journal of pediatrics. 2022 Sep 8:48(1):170. doi: 10.1186/s13052-022-01365-9. Epub 2022 Sep 8 [PubMed PMID: 36076277]
Glaeser AB, Diniz BL, Santos AS, Guaraná BB, Muniz VF, Carlotto BS, Everling EM, Noguchi PY, Garcia AR, Miola J, Riegel M, Mergener R, Gazzola Zen PR, Machado Rosa RF. A child with cat-eye syndrome and oculo-auriculo-vertebral spectrum phenotype: A discussion around molecular cytogenetic findings. European journal of medical genetics. 2021 Nov:64(11):104319. doi: 10.1016/j.ejmg.2021.104319. Epub 2021 Aug 30 [PubMed PMID: 34474176]
Liehr T, Fleischer N, Al-Rikabi A. Next-generation phenotyping in cat-eye syndrome based on computer-aided facial dysmorphology analysis of normal photographs. Molecular genetics & genomic medicine. 2021 Oct:9(10):e1785. doi: 10.1002/mgg3.1785. Epub 2021 Aug 25 [PubMed PMID: 34432367]
Katz B, Enright J, Couch S, Harocopos G, Lee AR. Atypical presentation of Cat Eye Syndrome in an infant with Peters anomaly and microphthalmia with cyst. Ophthalmic genetics. 2020 Dec:41(6):645-649. doi: 10.1080/13816810.2020.1814346. Epub 2020 Aug 31 [PubMed PMID: 32865081]
McTaggart KE, Budarf ML, Driscoll DA, Emanuel BS, Ferreira P, McDermid HE. Cat eye syndrome chromosome breakpoint clustering: identification of two intervals also associated with 22q11 deletion syndrome breakpoints. Cytogenetics and cell genetics. 1998:81(3-4):222-8 [PubMed PMID: 9730608]
Karcaaltincaba D, Ceylaner S, Ceylaner G, Dalkilic S, Karli-Oguz K, Kandemir O. Partial trisomy due to a de novo duplication 22q11.1-22q13.1: a cat-eye syndrome variant with brain anomalies. Genetic counseling (Geneva, Switzerland). 2010:21(1):19-24 [PubMed PMID: 20420025]
Knijnenburg J, van Bever Y, Hulsman LO, van Kempen CA, Bolman GM, van Loon RL, Beverloo HB, van Zutven LJ. A 600 kb triplication in the cat eye syndrome critical region causes anorectal, renal and preauricular anomalies in a three-generation family. European journal of human genetics : EJHG. 2012 Sep:20(9):986-9. doi: 10.1038/ejhg.2012.43. Epub 2012 Mar 7 [PubMed PMID: 22395867]
Mears AJ, Duncan AM, Budarf ML, Emanuel BS, Sellinger B, Siegel-Bartelt J, Greenberg CR, McDermid HE. Molecular characterization of the marker chromosome associated with cat eye syndrome. American journal of human genetics. 1994 Jul:55(1):134-42 [PubMed PMID: 7912885]
McDermid HE, Morrow BE. Genomic disorders on 22q11. American journal of human genetics. 2002 May:70(5):1077-88 [PubMed PMID: 11925570]
Giampietro PF. 50 Years Ago in TheJournalofPediatrics: "CatEye Syndrome" and the Unraveling of the 22q.11.2 Genomic Region. The Journal of pediatrics. 2020 Apr:219():88. doi: 10.1016/j.jpeds.2019.10.005. Epub [PubMed PMID: 32204809]
Dicipulo R, Norton KA, Fairbridge NA, Kibalnyk Y, Fox SC, Hornberger LK, McDermid HE. Cecr2 mutant mice as a model for human cat eye syndrome. Scientific reports. 2021 Feb 4:11(1):3111. doi: 10.1038/s41598-021-82556-y. Epub 2021 Feb 4 [PubMed PMID: 33542446]
Hernández-Medrano C, Hidalgo-Bravo A, Villanueva-Mendoza C, Bautista-Tirado T, Apam-Garduño D. Mosaic cat eye syndrome in a child with unilateral iris coloboma. Ophthalmic genetics. 2021 Feb:42(1):84-87. doi: 10.1080/13816810.2020.1839918. Epub 2020 Dec 1 [PubMed PMID: 33465332]
Sharma D, Murki S, Pratap T, Vasikarla M. Cat eye syndrome. BMJ case reports. 2014 May 19:2014():. doi: 10.1136/bcr-2014-203923. Epub 2014 May 19 [PubMed PMID: 24842361]
Level 3 (low-level) evidenceAlSubaihin A, VanderMeulen J, Harris K, Duck J, McCready E. Müllerian Agenesis in Cat Eye Syndrome and 22q11 Chromosome Abnormalities: A Case Report and Literature Review. Journal of pediatric and adolescent gynecology. 2018 Apr:31(2):158-161. doi: 10.1016/j.jpag.2017.09.004. Epub 2017 Sep 14 [PubMed PMID: 28919146]
Level 3 (low-level) evidenceXue H, Chen X, Lin M, Lin N, Huang H, Yu A, Xu L. Prenatal diagnosis and molecular cytogenetic identification of small supernumerary marker chromosomes: analysis of three prenatal cases using chromosome microarray analysis. Aging. 2020 Dec 9:13(2):2135-2148. doi: 10.18632/aging.202220. Epub 2020 Dec 9 [PubMed PMID: 33318309]
Level 3 (low-level) evidenceWu X, An G, He D, Shen Q, Cai M, Huang H, Lin Y, Xu L. [Prenatal diagnosis and clinical analysis of two fetuses with Cat-eye syndrome]. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics. 2019 May 10:36(5):498-501. doi: 10.3760/cma.j.issn.1003-9406.2019.05.021. Epub [PubMed PMID: 31030443]
Alamer L, Bassant S, Alhazmi R, Alzahrani M. Rare otologic presentation of cat eye syndrome. Annals of Saudi medicine. 2019 Nov-Dec:39(6):441-443. doi: 10.5144/0256-4947.2019.441. Epub 2019 Dec 5 [PubMed PMID: 31804144]
Bremond-Gignac D, Morin G, Jedraszak G, Receveur A, Rochette J, Copin H. [Cat eye syndrome]. Journal francais d'ophtalmologie. 2015 Apr:38(4):368-9. doi: 10.1016/j.jfo.2014.09.016. Epub 2015 Mar 20 [PubMed PMID: 25799388]
Seehase M, Houthuizen P, Collins JJ, Zimmermann LJ, Kramer BW. Propofol administration to the fetal-maternal unit reduces cardiac oxidative stress in preterm lambs subjected to prenatal asphyxia and cardiac arrest. Pediatric research. 2016 May:79(5):748-53. doi: 10.1038/pr.2016.10. Epub 2016 Jan 13 [PubMed PMID: 26761124]
Kharasch ED. Biotransformation of sevoflurane. Anesthesia and analgesia. 1995 Dec:81(6 Suppl):S27-38 [PubMed PMID: 7486145]
Brown DD, Dormois JC, Abraham GN, Lewis K, Dixon K. Effect of furosemide on the renal excretion of digoxin. Clinical pharmacology and therapeutics. 1976 Oct:20(4):395-400 [PubMed PMID: 975715]
. Erratum to "Cat eye syndrome: Clinical, cytogenetics and familial findings in a large cohort of 43 patients highlighting the importance of congenital heart disease and inherited cases". American journal of medical genetics. Part A. 2024 Nov:194(11):e63807. doi: 10.1002/ajmg.a.63807. Epub 2024 Jul 4 [PubMed PMID: 38963279]
Level 3 (low-level) evidenceWalter A, Calite E, Berg C, Gembruch U, Müller A, Geipel A. Prenatal diagnosis of fetal growth restriction with polyhydramnios, etiology and impact on postnatal outcome. Scientific reports. 2022 Jan 10:12(1):415. doi: 10.1038/s41598-021-04371-9. Epub 2022 Jan 10 [PubMed PMID: 35013541]
Kozlova A, DeMaria LN, Tran AQ, North VS, Belinsky I. Congenital Eyelid Imbrication and Floppy Eyelid Syndrome in a Patient With Cat Eye Syndrome. Ophthalmic plastic and reconstructive surgery. 2022 Mar-Apr 01:38(2):e36-e38. doi: 10.1097/IOP.0000000000002074. Epub [PubMed PMID: 34652311]
Chen CP, Ko TM, Chen YY, Su JW, Wang W. Prenatal diagnosis and molecular cytogenetic characterization of mosaicism for a small supernumerary marker chromosome derived from chromosome 22 associated with cat eye syndrome. Gene. 2013 Sep 15:527(1):384-8. doi: 10.1016/j.gene.2013.05.061. Epub 2013 Jun 6 [PubMed PMID: 23747353]
Makarov IA, Gavrilina SB, Belozerov BG. [Cat-eye syndrome (a psychiatric aspect)]. Zhurnal nevrologii i psikhiatrii imeni S.S. Korsakova. 2019:119(11):60-64. doi: 10.17116/jnevro201911911160. Epub [PubMed PMID: 31851174]
Berends MJ,Tan-Sindhunata G,Leegte B,van Essen AJ, Phenotypic variability of Cat-Eye syndrome. Genetic counseling (Geneva, Switzerland). 2001; [PubMed PMID: 11332976]
Busse T, Graham JM Jr, Feldman G, Perin J, Catherwood A, Knowlton R, Rappaport EF, Emanuel B, Driscoll DA, Saitta SC. High-Resolution genomic arrays identify CNVs that phenocopy the chromosome 22q11.2 deletion syndrome. Human mutation. 2011 Jan:32(1):91-7. doi: 10.1002/humu.21395. Epub [PubMed PMID: 21120947]
Ng FYC, Song HJJMD, Tan BKJ, Teo CB, Wong ETY, Boey PY, Cheng CY. Bidirectional association between glaucoma and chronic kidney disease: A systematic review and meta-analysis. EClinicalMedicine. 2022 Jul:49():101498. doi: 10.1016/j.eclinm.2022.101498. Epub 2022 Jun 8 [PubMed PMID: 35747173]
Level 1 (high-level) evidenceShi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nature reviews. Drug discovery. 2017 Feb:16(2):115-130. doi: 10.1038/nrd.2016.245. Epub 2016 Dec 16 [PubMed PMID: 27980341]
Cuesta AM, Gallardo-Vara E, Casado-Vela J, Recio-Poveda L, Botella LM, Albiñana V. The Role of Propranolol as a Repurposed Drug in Rare Vascular Diseases. International journal of molecular sciences. 2022 Apr 11:23(8):. doi: 10.3390/ijms23084217. Epub 2022 Apr 11 [PubMed PMID: 35457036]
Shen Y, Kong H, Zeng H, Wu Q, Chen J, Zhou D, Zhang J, Ge Y, Ding F. [Cytogenetic and molecular genetic analysis of the amniotic fluid cells of a fetus with pseudodicentric isochromosome 22 resulting in partial tetraploidy of 22q]. Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics. 2018 Apr 10:35(2):272-275. doi: 10.3760/cma.j.issn.1003-9406.2018.02.029. Epub [PubMed PMID: 29653009]
Jedraszak G, Braun K, Receveur A, Decamp M, Andrieux J, Rabbind Singh A, Copin H, Bremond-Gignac D, Mathieu M, Rochette J, Morin G. Growth hormone deficiency and pituitary malformation in a recurrent Cat-Eye syndrome: a family report. Annales d'endocrinologie. 2015 Oct:76(5):629-34. doi: 10.1016/j.ando.2015.02.002. Epub 2015 Oct 27 [PubMed PMID: 26518262]
Kohl S, Avni FE, Boor P, Capone V, Clapp WL, De Palma D, Harris T, Heidet L, Hilger AC, Liapis H, Lilien M, Manzoni G, Montini G, Negrisolo S, Pierrat MJ, Raes A, Reutter H, Schreuder MF, Weber S, Winyard PJD, Woolf AS, Schaefer F, Liebau MC. Definition, diagnosis and clinical management of non-obstructive kidney dysplasia: a consensus statement by the ERKNet Working Group on Kidney Malformations. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2022 Nov 23:37(12):2351-2362. doi: 10.1093/ndt/gfac207. Epub [PubMed PMID: 35772019]
Level 3 (low-level) evidenceUrsea R, Silverman RH. Anterior-segment imaging for assessment of glaucoma. Expert review of ophthalmology. 2010 Feb 1:5(1):59-74 [PubMed PMID: 20305726]
Evangelidou P, Sismani C, Ioannides M, Christodoulou C, Koumbaris G, Kallikas I, Georgiou I, Velissariou V, Patsalis PC. Clinical application of whole-genome array CGH during prenatal diagnosis: Study of 25 selected pregnancies with abnormal ultrasound findings or apparently balanced structural aberrations. Molecular cytogenetics. 2010 Nov 26:3():24. doi: 10.1186/1755-8166-3-24. Epub 2010 Nov 26 [PubMed PMID: 21110858]
Villagomez AN, Muñoz FM, Peterson RL, Colbert AM, Gladstone M, MacDonald B, Wilson R, Fairlie L, Gerner GJ, Patterson J, Boghossian NS, Burton VJ, Cortés M, Katikaneni LD, Larson JCG, Angulo AS, Joshi J, Nesin M, Padula MA, Kochhar S, Connery AK, Brighton Collaboration Neurodevelopmental Delay Working Group. Neurodevelopmental delay: Case definition & guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine. 2019 Dec 10:37(52):7623-7641. doi: 10.1016/j.vaccine.2019.05.027. Epub [PubMed PMID: 31783983]
Level 3 (low-level) evidenceIwai N, Fumino S. Surgical treatment of anorectal malformations. Surgery today. 2013 Sep:43(9):955-62. doi: 10.1007/s00595-012-0435-y. Epub 2012 Nov 30 [PubMed PMID: 23196862]
Arslan FN, Dogan DG, Canaloglu SK, Baysal SG, Buyukavci R, Buyukavci MA. Effects of early physical therapy on motor development in children with Down syndrome. Northern clinics of Istanbul. 2022:9(2):156-161. doi: 10.14744/nci.2020.90001. Epub 2022 Apr 18 [PubMed PMID: 35582517]
Melo C, Gama-de-Sousa S, Almeida F, Rendeiro P, Tavares P, Cardoso H, Carvalho S. Cat eye syndrome and growth hormone deficiency with pituitary anomalies: a case report and review of the literature. Gene. 2013 Oct 15:529(1):186-9. doi: 10.1016/j.gene.2013.07.031. Epub 2013 Aug 6 [PubMed PMID: 23928108]
Level 3 (low-level) evidenceBrugts JJ, Cuypers JA, Polak P, Ouhlous M, van de Woestijne P, Wessels M, Roos-Hesselink J. Quadricuspid aortic valve and anomalous systemic venous connection in a patient with cat-eye syndrome. Circulation. 2015 Mar 31:131(13):1225-7. doi: 10.1161/CIRCULATIONAHA.114.013290. Epub [PubMed PMID: 25825400]
Torti EE, Braddock SR, Bernreuter K, Batanian JR. Oculo-auriculo-vertebral spectrum, cat eye, and distal 22q11 microdeletion syndromes: a unique double rearrangement. American journal of medical genetics. Part A. 2013 Aug:161A(8):1992-8. doi: 10.1002/ajmg.a.35918. Epub [PubMed PMID: 23894059]
Quintero-Rivera F, Martinez-Agosto JA. Hemifacial microsomia in cat-eye syndrome: 22q11.1-q11.21 as candidate loci for facial symmetry. American journal of medical genetics. Part A. 2013 Aug:161A(8):1985-91. doi: 10.1002/ajmg.a.35895. Epub 2013 Jun 21 [PubMed PMID: 23794175]